Command Palette
Search for a command to run...
实现高选择性底物设计,MIT 联手哈佛用生成式 AI 发现全新蛋白酶切割模式

在生命体错综复杂的生化反应网络中,蛋白酶能够特异性地切割肽键,从而精准调控从凝血、组织修复到免疫应答乃至癌症演进等一系列关键生命过程。一旦其功能失常,往往直接导致多种重大疾病的发生与发展。因此,揭示蛋白酶的作用机制并精准调控其活性,不仅是基础生命科学的核心课题,也是开发新型诊疗手段的重要突破口。
实现这一目标的关键,在于找到与之高度「匹配」的肽底物——它们既能作为分子探针追踪酶活动态,也可被设计成抑制剂阻断异常活性,甚至能在药物递送系统中充当「条件激活开关」,实现靶向治疗。
然而,设计出既能被目标蛋白酶快速切割又具有高选择性(仅被该酶识别,避免与其他蛋白酶交叉反应)的肽底物,一直是科学界的重大挑战。这一难题根植于蛋白酶与底物之间复杂的生化互作:为适应多样生理功能,蛋白酶进化出广泛的切割特异性,其活性位点需与肽底物(通常长约十个氨基酸)精细结合。仅就 10 个氨基酸的合成肽(synthetic peptides)而言,使用常见的 20 种天然氨基酸,理论序列组合可达约 20¹⁰(近 10¹³)种,构成一个近乎无穷的探索空间。更复杂的是,功能相近的蛋白酶往往源自共同祖先,活性位点结构相似,极易发生「交叉识别」,这使得从海量可能性中筛选出高特异性底物变得尤为困难。
为突破这一瓶颈,研究人员已进行诸多尝试。传统方法多依赖已知的切割位点或天然蛋白质的酶切信息,效率较低,难以获得理想的人工底物;基于化学生物学知识的理性设计,则通常过程繁琐、通量有限,且多为针对单一蛋白酶,难以规模化应用。近年来,高通量筛选技术虽在一定程度上提升了效率,仍存在操作复杂、成本较高的限制;而现有计算预测方法大多仅能判断「是否切割」,无法准确排序切割效率,因此难以满足深度机理研究与工程应用的需求。
在此背景下,麻省理工学院与哈佛大学联合提出了基于人工智能的端到端设计流程 CleaveNet,通过预测模型与生成模型的协同工作,旨在彻底改变蛋白酶底物设计的现有范式,为相关基础研究与生物医药开发提供全新的解决方案。

论文地址:
https://www.nature.com/articles/s41467-025-67226-1
关注公众号,后台回复「CleaveNet」获取完整 PDF
更多 AI 前沿论文:
多实验场景数据集支撑,赋能 CleaveNet 模型泛化能力的跨场景验证
在开发与验证 CleaveNet 模型的过程中,该研究整合了两个在序列构成与实验方法上具有显著差异的数据集,以确保模型的可靠性与泛化能力。
研究人员使用的核心数据集来源于一项已公开的研究,该研究通过 mRNA 展示技术系统表征了一个包含约 18,500 条人工合成十肽的底物库对 18 种基质金属蛋白酶(MMPs)的切割活性。每个底物-蛋白酶组合均对应一个标准化的切割效率分数(Zₛₘ),用以量化相对切割强度。
为进一步确保评估的严谨性,避免因序列相似性导致的高估,研究人员对初始测试集进行了同源性过滤:计算每条测试序列与训练集中所有序列的最小莱文斯坦距离,并移除了 816 条距离小于 3 的、与训练集高度相似的序列。最终,研究人员获得了包含 2,901 条非重叠序列的「mRNA 展示测试集」,该子集在模型训练的任何阶段均未被使用,专用于内部性能验证。
为独立检验模型在面对截然不同的生物化学背景时的适应能力,该研究还引入了一个完全独立的分布外数据集,称为「荧光测试集」。该数据集包含 71 条长度不一(7-14 个氨基酸)的合成肽,其切割活性已通过基于荧光共振能量转移(FRET)的经典体外实验,针对 7 种重组 MMP 蛋白进行了验证。该数据集在合成肽长度分布、氨基酸组成以及最关键的实验检测原理上,均与基于 mRNA 展示技术生成的核心数据集存在本质不同。这种有意的差异设计为评估 CleaveNet 模型超越特定实验条件、捕捉普适性生化规律的能力提供了关键基准。
CleaveNet 形成预测-生成协同闭环
如下图所示,CleaveNet 的核心由两个功能互补且协同工作的计算模块构成:预测模块(CleaveNet Predictor)与生成模块(CleaveNet Generator),二者共同形成一个完整的「设计-评估」闭环。
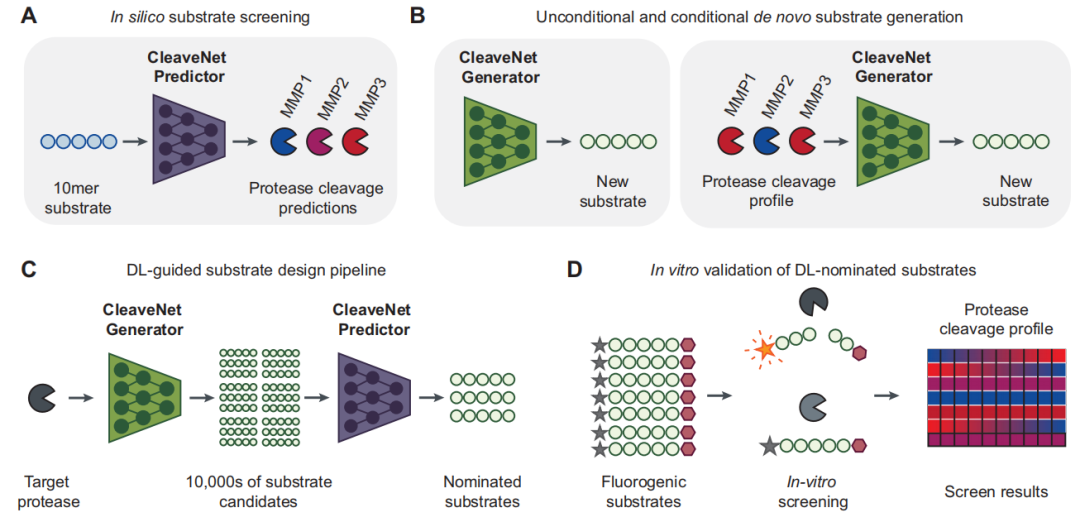
预测模块旨在解决从海量序列空间中快速、精准评估候选底物切割活性的问题。研究人员将其构建为一个多输出的「序列-功能」回归模型。具体而言,该模型以氨基酸序列作为输入,其核心任务是同时输出该序列对全部 18 种 MMPs 的预测切割分数(Ŵₛₘ),并同步估算每个预测的不确定性(σₛₘ)。
为实现更高的预测稳健性,该研究采用了模型集成策略:在 mRNA 展示训练集上独立训练了 5 个相同的预测模型,最终的预测分数为其输出均值,而预测的不确定性则由这 5 个结果的标准差量化。此外,通过设定一个可调的阈值(Zₜ),该模型可将连续的预测分数便捷地转换为「切割」或「未切割」的二元判断,从而服务于不同的筛选场景。
在构建预测模型时,该研究对序列建模中两种主流架构——双向长短期记忆网络与 Transformer——进行了系统对比。前者擅于捕捉序列中的顺序依赖关系,而后者凭借其注意力机制能全局建模氨基酸之间的相互作用,是当前蛋白质语言表征的主流选择。基于在更大规模、更高多样性数据上展现的潜力,研究人员最终选择了 Transformer 架构作为 CleaveNet Predictor 的基础。
生成模块的目标则是实现候选底物的自动化、智能化设计。该研究训练了一个基于自回归 Transformer 的生成模型,使其能够学习 mRNA 展示数据集中蕴含的、普适性的 MMP 切割偏好。该模型无需任何额外条件输入,即可生成大量新颖且合理的肽序列。
为了科学评估生成模型的价值而非简单地复现随机性,研究人员建立了一个强有力的基线方法,称为「位点独立对照」。该方法仅统计训练数据中各氨基酸位置上的独立分布,并据此进行随机采样以生成序列。通过对比 CleaveNet 生成序列与这种基线序列在多个维度上的差异,能够清晰地辨别模型所学习的、超越简单统计关联的复杂生化规律。
预测模块与生成模块的紧密协作,使得研究人员能够首先生成多样化的候选库,继而对其进行高效、精准的虚拟筛选,从而为后续的实验验证提供了强有力的计算驱动引擎。
CleaveNet 可实现选择性精准调控
在完成模型构建后,该研究对 CleaveNet 的性能进行了多层次、系统性的实验验证,其结果充分证明了该流程在预测准确性、生成合理性及实际应用有效性方面的突出价值。
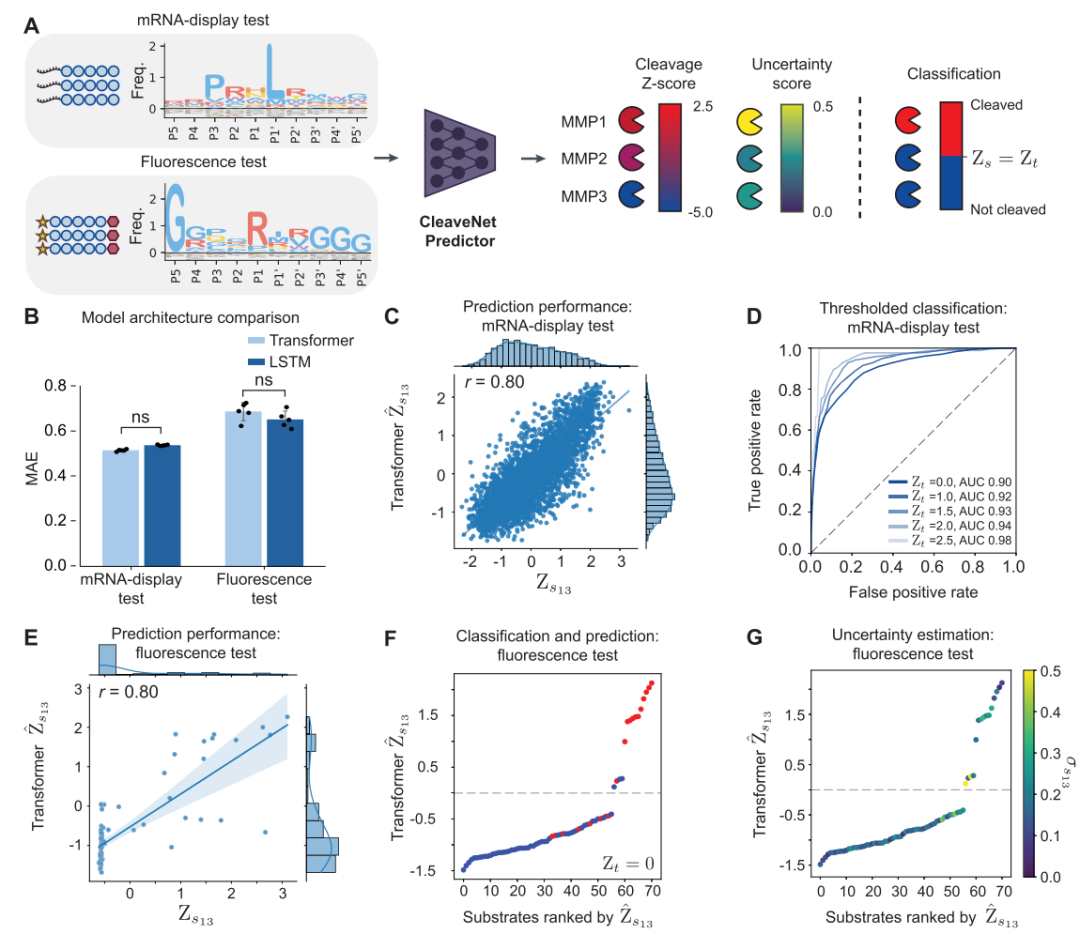
首先,CleaveNet Predictor 在内部与外部测试集上均展现出卓越的预测能力。在从未参与训练的同源过滤测试集(mRNA 展示测试集)上,模型对 MMP13 的预测分数(Ŵₛₘ)与实验测得的标准化 Z 分数(Zₛₘ)呈现出强相关性(皮尔逊相关系数 r = 0.80)。当将连续的预测值转换为「切割/未切割」的二元分类时,其性能同样稳健:通过绘制受试者工作特征(ROC)曲线并计算曲线下面积(AUC),研究人员发现模型在不同判定阈值下均保持高区分度,尤其在通用可信切割阈值(Zₜ=2.5)下,AUC 高达 0.98 。更严格的考验来自完全独立、实验方法迥异的荧光测试集。
尽管该数据集的序列长度、氨基酸组成及检测原理均与训练数据不同,模型预测的切割分数与实验值之间依然维持强正相关(针对 MMP13,r = 0.80),并能准确地将实验验证的「切割」与「未切割」序列区分开来。这有力地证实了 CleaveNet Predictor 不仅能够记忆训练数据模式,更能捕捉底物被蛋白酶切割的普适性生化规律,具备强大的泛化能力。
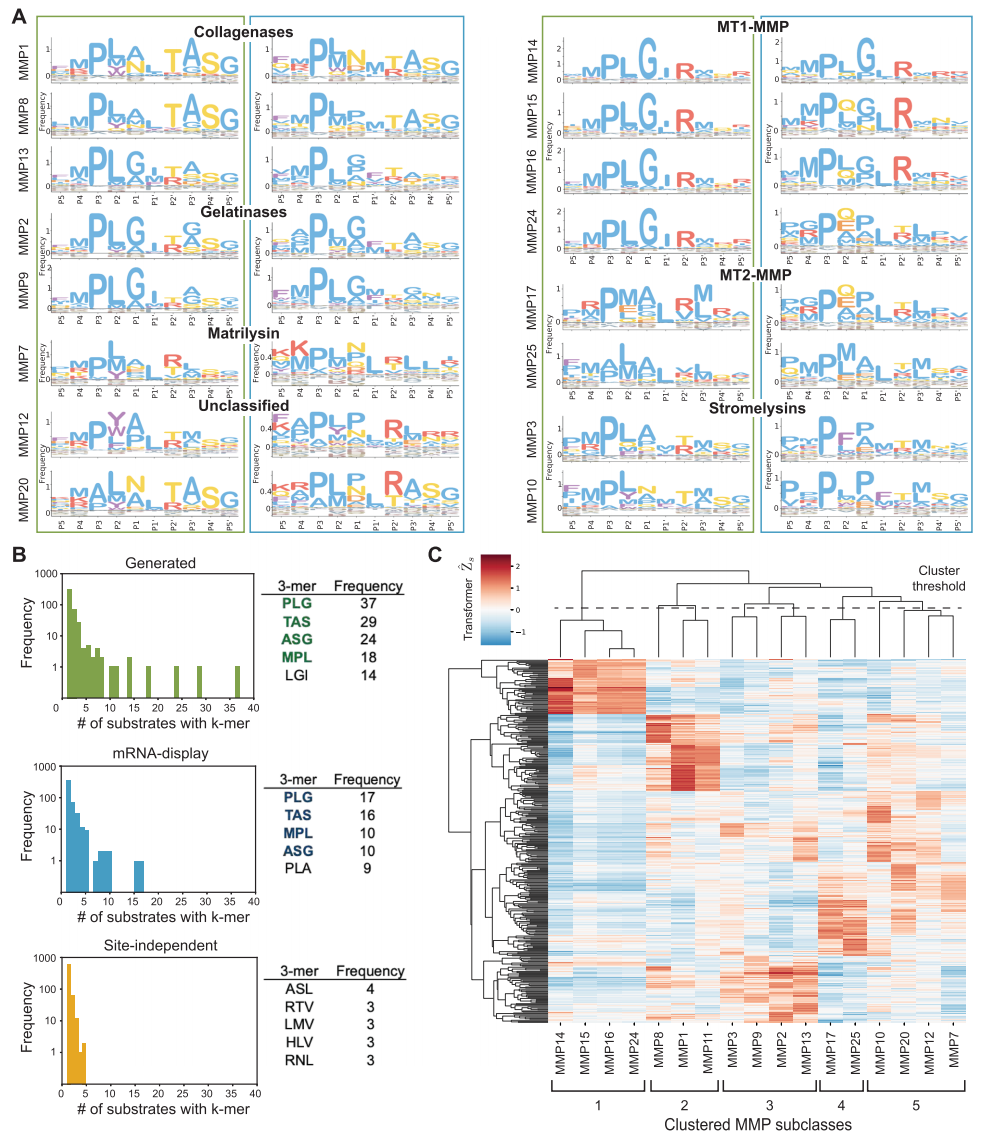
其次,研究人员对 CleaveNet Generator 生成序列的生物信息学分析揭示了其合理性与新颖性。与仅基于单氨基酸位置频率随机采样的「位点独立对照」序列相比,生成模型产生的序列更精准地复现了 MMP 家族经典的切割基序,并在关键的底物结合口袋区域呈现出与真实实验数据更为接近的氨基酸分布。更重要的是,生成序列在整体生物物理特性(如疏水性、电荷)上与真实数据集保持一致。然而,高质量的生成并非意味着对训练数据的简单复制。序列多样性分析显示,生成序列与训练集共享的独特长合成肽比例极低,表明模型避免了过拟合,能够探索训练数据未能覆盖的新序列空间。
进一步的功能聚类分析发现,针对不同 MMP 所生成的高评分底物,其预测的切割活性谱能够依据 MMP 催化结构域的系统发育关系自然聚类。这证明生成模型不仅学会了表观的序列模式,更内在地捕捉了蛋白酶进化上的功能分化信息,体现了其生成结果的生物学合理性。
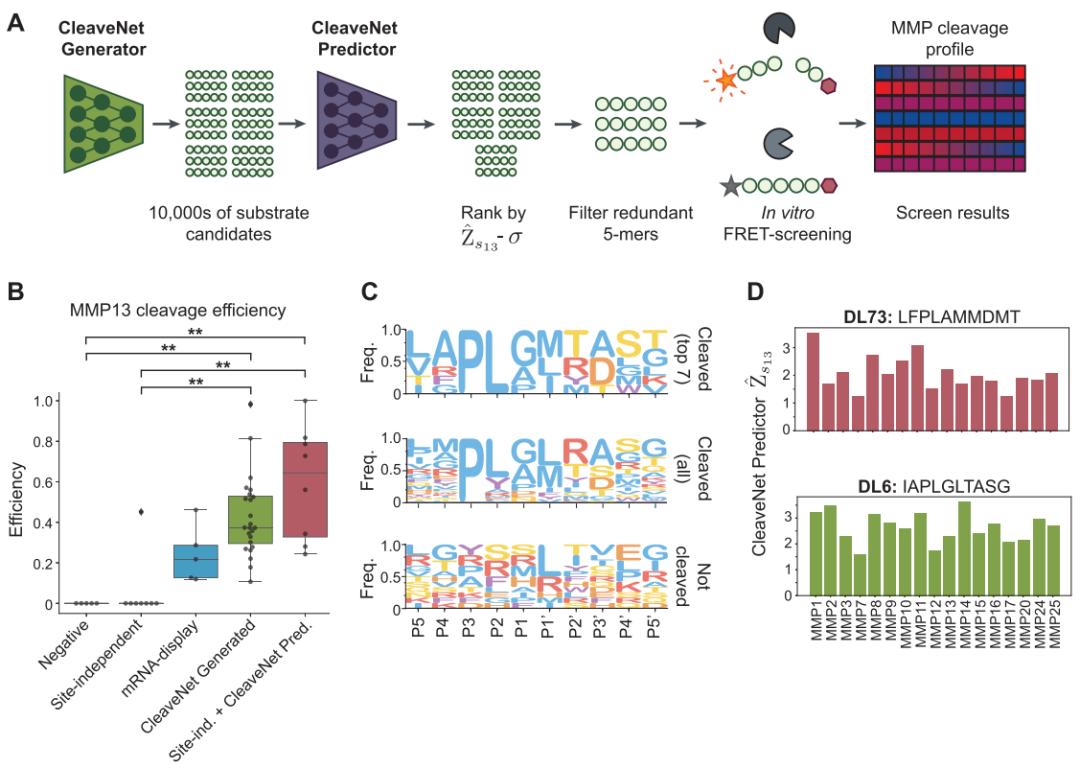
最终,所有计算设计的有效性都通过了体外生化实验的终极检验。研究人员合成了多组由 CleaveNet 设计、针对 MMP13 的候选底物,包括由生成模型直接产生的序列,以及经过预测模型筛选的序列。荧光共振能量转移(FRET)切割实验给出了令人信服的结果:所有 24 条由 CleaveNet 全流程设计的底物均被重组 MMP13 成功切割,命中率达到 100%,且其中值切割效率显著高于训练集中已知的高效阳性对照底物。这验证了该流程设计高效底物的能力。
为了展示流程在解决更具挑战性任务(如设计高选择性底物)方面的潜力,该研究进一步采用了条件生成策略,即在生成模型中指定「高 MMP13 选择性」作为目标。随后开展的大规模并行体外筛选(95 条设计底物对 12 种不同 MMP)结果表明,通过条件引导生成的底物,其切割活性显著偏向于 MMP13,实现了更高的选择性。
尤为引人注目的是,部分设计底物同时兼备了高切割效率与高选择性,这一在原始训练数据中极为罕见的优异组合,凸显了 CleaveNet 在探索全新、优质序列空间方面的强大能力。
综上所述,从精准的计算预测到合理的序列生成,再到确凿的湿实验验证,一系列环环相扣的结果共同表明,CleaveNet 构建了一个高效、可靠且功能强大的蛋白酶底物设计平台。该研究不仅为蛋白酶活性调控这一经典难题提供了创新的 AI 解决方案,也为未来的蛋白酶功能研究与相关药物开发奠定了崭新的方法论基础。
AI 驱动蛋白酶底物设计革新
CleaveNet 所引领的 AI 辅助蛋白酶底物设计技术,正在全球范围内推动生命科学与生物医药领域的创新。
美国华盛顿大学 David Baker 团队在 Science 上发表突破性研究,首次利用 AI 从头设计出具有复杂活性位点的丝氨酸水解酶——这是已知最大的酶家族之一。研究引入新型机器学习网络 PLACER,不仅成功设计出可高效催化酯水解反应的活性酶,还意外发现了五种全新的蛋白质折叠方式,显著扩展了该酶家族的结构多样性。
* 论文标题:Computational design of serine hydrolases
* 论文链接:
https://www.science.org/doi/10.1126/science.adu2454
此外,欧洲多所高校的联合团队基于 Transformer 架构,构建了能精准预测蛋白酶-底物相互作用的通用模型。该模型整合了全球多源蛋白酶切割数据,实现了跨物种底物序列的有效预测,其泛化能力已在细菌、病毒等多种病原体蛋白酶研究中得到验证,为开发抗感染药物提供了重要的序列设计依据。
可以预见,随着计算生物学、人工智能与合成生物学的持续交汇,蛋白酶底物设计将从一门兼具艺术与经验的科学,进化为一个高度理性化、工程化的研究领域。这不仅会加速新型药物、诊断工具和绿色生物催化剂的开发,更有可能帮助我们最终解码生命调控的底层逻辑,开启按需编程生命功能的新时代。








