Command Palette
Search for a command to run...
By Linking Gene Expression Data With Cell Morphology Images, Researchers at the Chinese University of Hong Kong and Others Have Developed a transcriptome-guided Diffusion Model to Accelerate Phenotypic Drug development.
Cell morphology is a core research area in single-cell biology. Its value lies in systematically analyzing the dynamic changes in cell morphology under genetic or drug perturbations through high-throughput image analysis. This research not only significantly improves the accuracy of compound mechanism of action (MOA) predictions but also enhances the accuracy of compound bioactivity assessments. Ultimately, it provides data support for key steps in phenotypic drug discovery, such as candidate compound screening and mechanism of action verification, effectively accelerating the R&D process.
However, observing and analyzing changes in cell morphology following genetic or drug perturbations is not a simple task. The number of compounds that can be screened exceeds millions, and the number of genes that can be edited is also in the tens of thousands. Using traditional experimental methods to verify each one individually is not only extremely inefficient but also incurs high time and financial costs. Although various computational methods have been proposed and applied to cell morphology prediction, their accuracy and fidelity still cannot meet the needs of practical research.
Specifically,The limitations of existing methods are mainly reflected in two points:First, the performance of advanced models, such as IMPA (IMage Perturbation Autoencoder), is highly dependent on known biological knowledge or specific datasets, resulting in weak generalization capabilities and lack of wide applicability. Second, cell morphology data is easily affected by experimental interference factors such as batch effects and well position effects, and exhibits a high level of noise, which makes it difficult to effectively capture the true cell morphological characteristics, which in turn directly affects the stability and reliability of the data and restricts the accuracy of subsequent analysis results.
To address the above challenges, researchers from the Chinese University of Hong Kong, Mohamed bin Zayed University of Artificial Intelligence and other institutions proposed a scalable transcriptome-guided diffusion model, MorphDiff.This model is specifically designed for high-fidelity simulation of cell morphology responses to perturbations. It is based on the Latent Diffusion Model (LDM) architecture and uses the L1000 gene expression profile as conditional input for denoising training.
The results of the study confirmed thatThe core advantage of MorphDiff is its ability to accurately generate cell morphologies under "unseen perturbation conditions".This capability provides dual key benefits: First, it helps researchers efficiently explore the vast space of phenotypic perturbation screening, significantly reducing reliance on large-scale field experiments, thereby reducing experimental costs and improving screening efficiency; second, it assists in elucidating the mechanisms of action of structurally diverse drug molecules, providing key support for compound mechanism validation. Therefore, MorphDiff can serve as a high-performance tool to accelerate phenotypic drug development.
The research results were published in Nature Communications under the title "Prediction of cellular morphology changes under perturbations with a transcriptome-guided diffusion model."
Research highlights:
* This study innovatively applies diffusion models to predict cell morphology for the first time, opening up new avenues and providing new tools for phenotypic drug development.
* Extensive benchmark tests demonstrate the effectiveness of MorphDiff, especially in MOA retrieval, where it can achieve comparable accuracy to ground truth morphology, outperforming baseline methods by 16.9% and 8% respectively.

Paper address:
https://www.nature.com/articles/s41467-025-63478-z
Follow the official account and reply "transcriptome-guided diffusion" to get the full PDF
More AI frontier papers:
https://hyper.ai/papers
Dataset: Large-scale multi-omics dataset to verify its effectiveness
To systematically verify the effectiveness and generalization of the MorphDiff model in predicting cell morphology under perturbation conditions,This study constructed a multi-cell line and multi-source dataset system based on the dual dimensions of "genetic perturbation-drug perturbation".For each sample in the experiment, two types of data were collected: an L1000 gene expression profile and a cell morphology image, forming paired data. The former served as the "molecular feature input," and the latter as the "phenotypic feature input." This method ensures the correlation between the perturbation signal at the genetic level and the phenotypic response at the morphological level, driven solely by the target perturbation. This eliminates interference from irrelevant variables such as cell line differences and experimental batches.
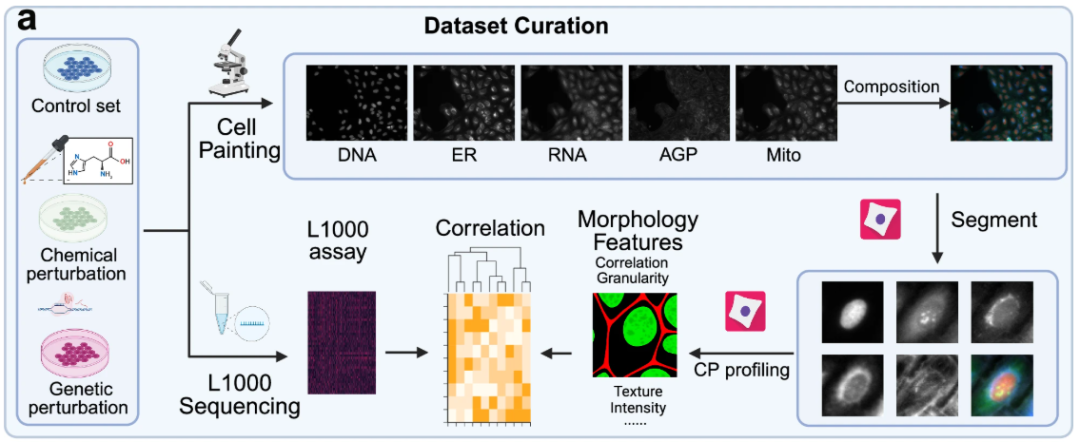
In terms of cell morphology image datasets,The study used three large-scale cell morphology image datasets for model training, evaluation, and analysis: one genetic perturbation dataset and two drug perturbation datasets. The genetic perturbation dataset, derived from the JUMP dataset based on the U2OS cell line, contains 130 genetic perturbations. The drug perturbation datasets, derived from the CDRP dataset based on the U2OS cell line and the LINCS dataset based on the A549 cell line, contain 1,028 drug perturbations and 61 drug perturbations, respectively.
All three cell morphology image datasets were preprocessed and segmented. CellProfiler 4.2.5 was used to segment the bulk cell plate images into single-cell images for more granular analysis.The cell morphology images obtained by Cell Painting technology contain 5 core channels.That is, DNA (nucleus), RNA (nucleolus and cytoplasm), ER (endoplasmic reticulum), AGP (Golgi apparatus/cell membrane/actin skeleton) and Mito (mitochondria).
besides,The experiment further planned the L1000 dataset of "no corresponding morphological images".This dataset is mainly used to further explore the application of the model in the scenario of "only obtaining gene expression data", and can lay the foundation for subsequent mechanism verification, drug screening, etc. to provide "data-driven" hypotheses.
Model architecture and methods: linking gene expression data with cell morphology images
The core goal of MorphDiff is to achieve end-to-end accurate mapping from L1000 gene expression profiles to cell morphology images through a transcriptome-guided latent diffusion model framework.Simply put, it is to design and train a model, which is like a "bridge" - input the L1000 gene expression data corresponding to a certain "disturbance", and then output the morphological image or perturbation morphology of the cell under this perturbation.
The core of the MorphDiff model consists of two major modules:Morphology Variational Autoencoder (MVAE) and Latent Diffusion Model (LDM), as shown in Figure b below.
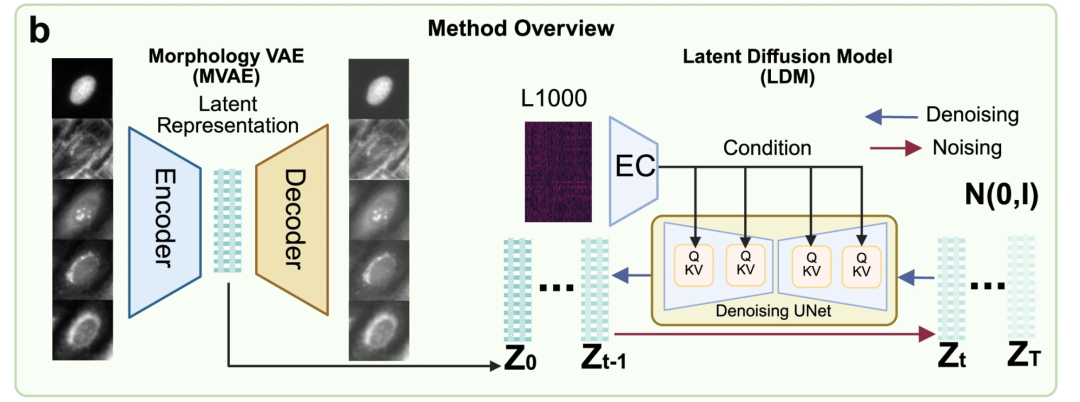
Among them, MVAE is the "image compression engine" of the model, which is responsible for converting multi-channel, high-resolution cell morphology images into low-dimensional, interpretable potential representations.This approach addresses the high computational cost and unstable training associated with directly training diffusion models on high-dimensional images. Structurally, the MVAE consists of two parts: an encoder and a decoder. The encoder compresses complex 5-channel morphological images into a simpler low-dimensional representation; the decoder restores the original morphological image from this low-dimensional representation.
LDM is mainly responsible for binding "gene expression" and "compressed morphological features", helping the model complete the relationship "from genes to morphological features". LDM involves a denoising process and a denoising process. The denoising process gradually adds Gaussian noise to the compressed morphological features until they are completely random. The denoising process allows the model to gradually restore the random noise to the original morphological features, given the known expression of L1000 genes. The model uses a U-Net network architecture and incorporates an attention mechanism to more accurately link key genetic and morphological information.
Figure c below demonstrates two applications of the pre-trained MorphDiff model: G2I and I2I. The former denoises the corresponding cell morphology image from a random noise distribution, conditioned on L1000 gene expression, to generate the corresponding cell morphology image. The latter, conditioned on L1000 gene expression under a specific perturbation, transforms the morphology image from the control cell morphology to the predicted perturbed morphology image, achieving "normal to perturbed morphology" prediction capabilities.
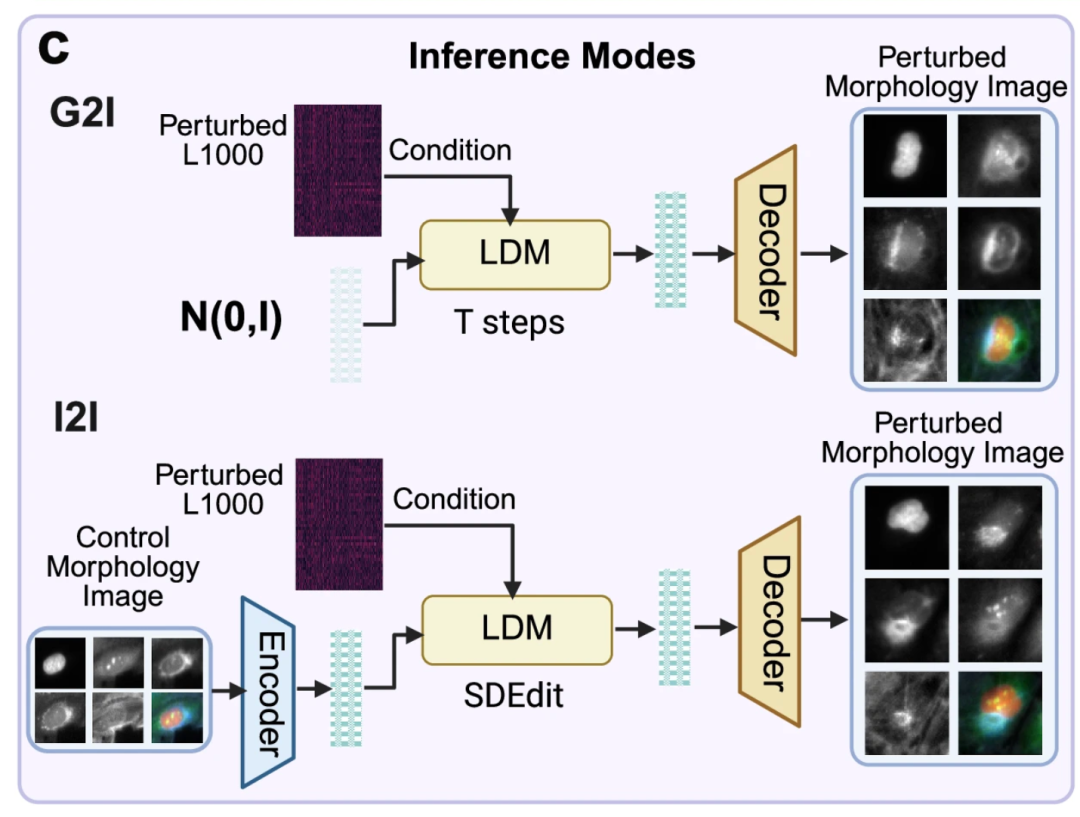
Figure d below shows the value of the MorphDiff model in practical applications.First, the model is able to predict cell morphological changes caused by "unseen perturbations" that were not encountered during training.This allows researchers to simulate the state of cell changes under the disturbance of new drugs by computers without having to conduct physical experiments, thus helping them to explore more possibilities quickly and at low cost; secondly,The model framework combines tools such as CellProfiler and DeepProfiler.It can help identify the MOA of drugs, thereby promoting the development of phenotypic drugs.
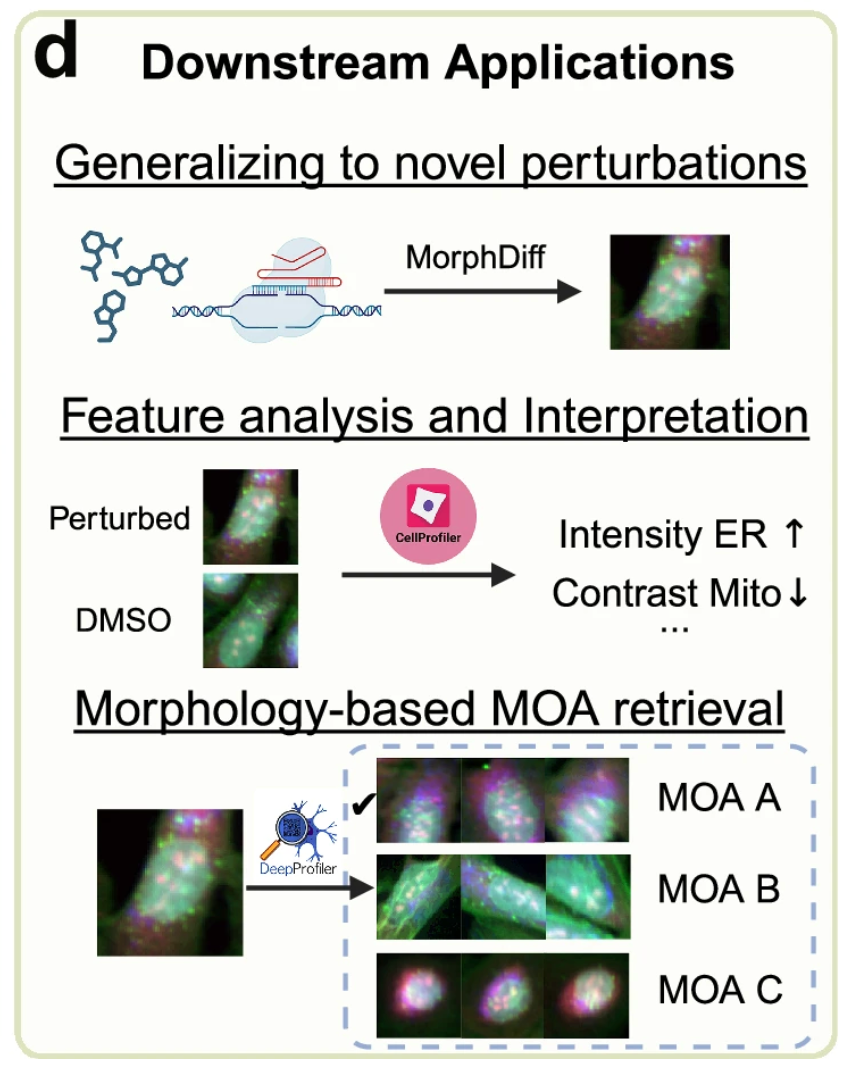
The researchers say MorphDiff is currently the only tool that supports the generation of morphological images from gene expression and the conversion from unperturbed morphology to perturbed morphology.
Experimental results: Performance surpasses IMPA, accelerating phenotypic drug development
In order to verify the effectiveness of the MorphDiff model, researchers designed a series of experiments for different purposes.Through experimental comparison with advanced tools, the effectiveness and practicality of MorphDiff are systematically verified.
First, the experiments verified the genetic perturbation predictions.The researchers conducted benchmark tests on the JUMP OOD dataset and compared them with multiple baseline methods, including MorphNet, DMIT (Disentanglement for Multi-mapping Image-to-Image Translation), DRIT++ (Disentangled Representation for Image-to-Image Translation), StarGANv1, IMPA, VQGAN (Vector Quantized Generative Adversarial Network), and MDTv2 (Masked Diffusion Transformers).
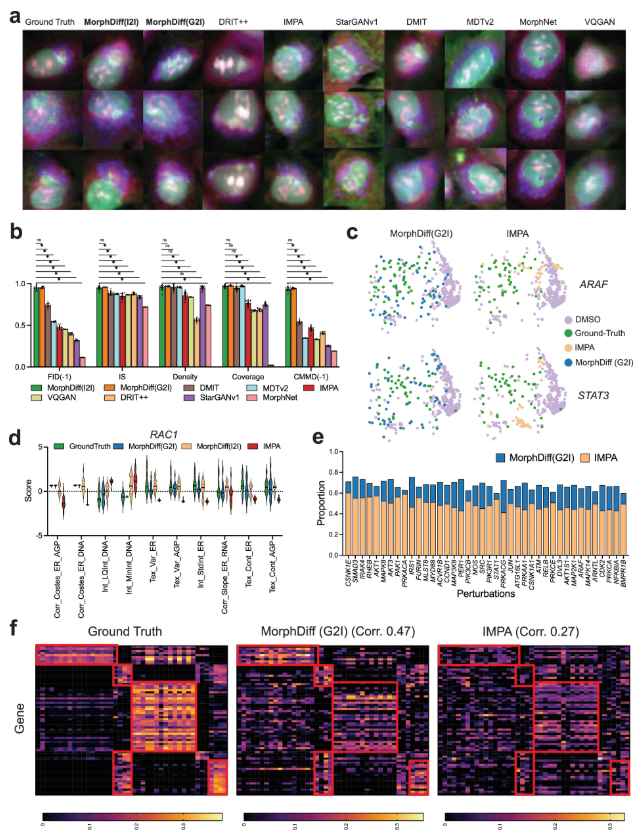
The results show thatThe results generated by the two modes of MorphDiff are closer to the real baseline in terms of visual quality and structural morphology.In terms of quantitative metrics, including FID, IS (Inception Score), CMMD, density, and coverage, both MorphDiff modes outperformed baseline methods in generalization, fidelity, and diversity, achieving higher output quality. In predicting cell morphology changes, MorphDiff (G2I) outputs are more diverse and closer to the true baseline; while the features generated by MorphDiff (I2I) exhibit higher overlap with the true baseline features, indicating that its prediction accuracy is significantly consistent with the true perturbation morphology.
Afterwards, experiments were conducted to verify drug perturbation prediction. The researchers first benchmarked all the methods using the CDRP OOD dataset.MorphDiff achieves impressive performance compared to other baseline methods in most metrics, demonstrating its more powerful and stable comprehensive generation capabilities.The researchers then went on to perform a more rigorous evaluation of the model on the LINCS dataset, further investigating the response of cell morphology to small molecule compounds by comparing the generated results with CellProfiler features.
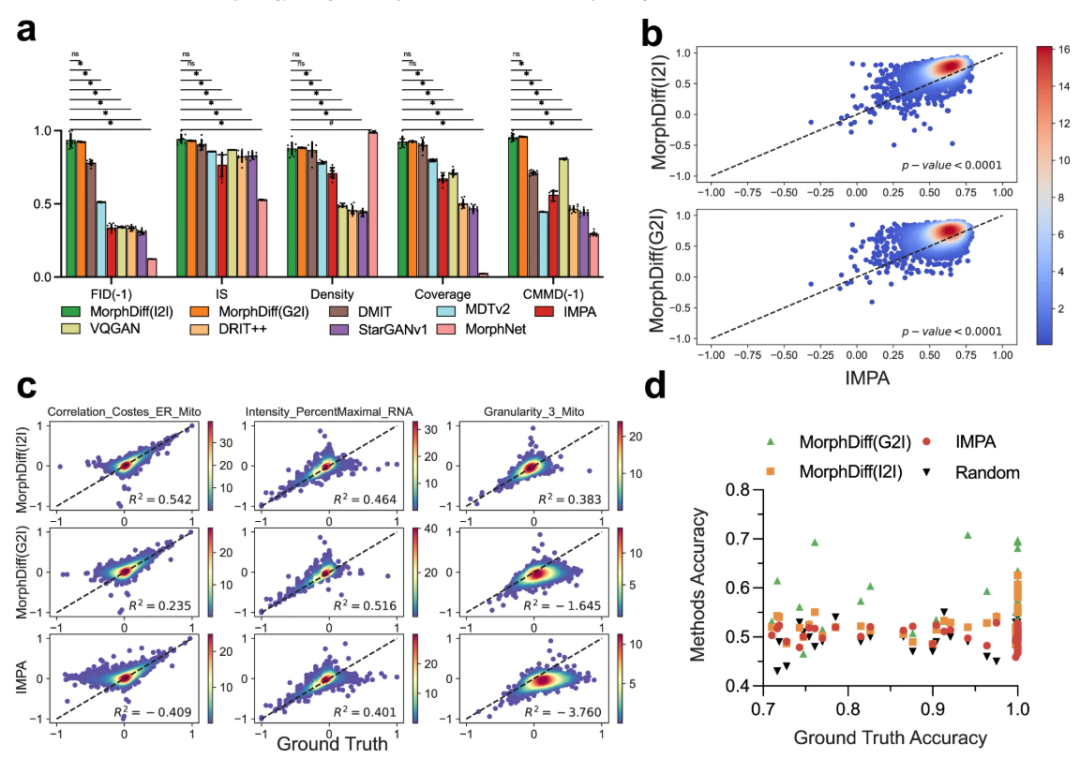
The results show that both modes of MorphDiff significantly outperform the baseline.Under G2I, 87.61 TP3T samples scored greater than 0.5, and 16.21 exceeded 0.8. Under I2I, 891 TP3T samples scored greater than 0.5, and 27.21 exceeded 0.8. In comparison, 78.31 TP3T samples from IMPA scored greater than 0.5, but none exceeded 0.8. The same analysis on the CDRP OOD dataset and the LINCS leave-one-out dataset revealed that both MorphDiff models outperformed the baseline method, with p-values less than 0.0001, demonstrating the generalizability of the method.
In DeepProfiler embedding analysis,MorphDiff (G2I) can most reliably and accurately capture perturbation-specific cell morphological patterns.At the pharmaceutical level,The output generated by the G2I pattern has higher perturbation specificity than that of I2I. These demonstrations further illustrate the potential of MorphDiff in drug screening.
Finally, the experiment also verified the capabilities of MorphDiff in drug development. The researchers selected the CDRP Target_MOA dataset to benchmark MorphDiff's two application modes and IMPA.
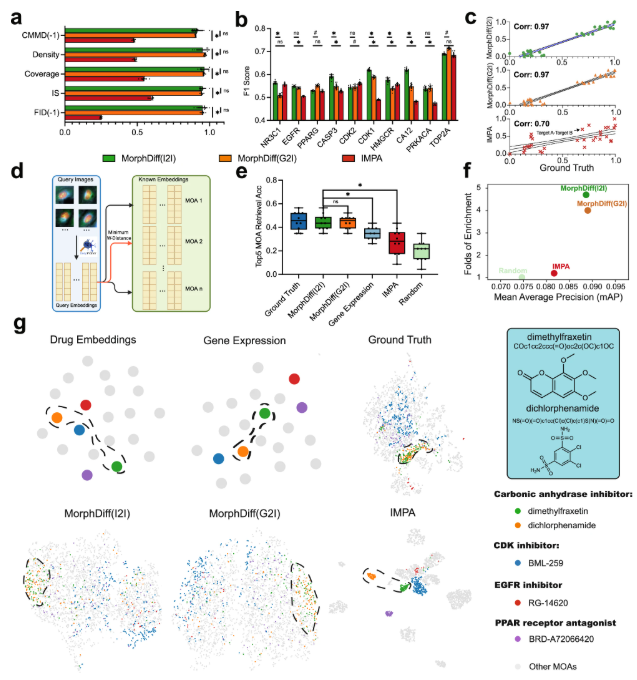
The results show that the perturbation morphology generated by MorphDiff is highly consistent with the true baseline.This demonstrated its ability to capture target-related diversity and effectively learn the complex relationship between drug perturbations and morphology. In the MOA retrieval task, the average accuracy of results generated by MorphDiff was 16.9% higher than that of IMPA-based and 8% higher than that of gene expression-based retrieval, respectively. Furthermore, experiments demonstrated that MorphDiff can discover that cell morphology contains complementary information and can identify drugs with the same MOA but different structures, which can help accelerate phenotypic drug development.
Silicon-based simulations become mainstream, accelerating downstream drug development
Phenotypic drug discovery differs from target-based drug discovery in that it explores and develops drugs by observing their effects on overall biological systems or cellular phenotypes. This approach offers significant advantages in discovering new mechanisms of action and targets, and in treating complex diseases. Numerous laboratories and research institutions are actively pursuing this topic, integrating computational technology with biomedicine to forge a new chapter in phenotypic drug discovery.
As mentioned in the paper, the IMPA model is a deep generative model proposed by a team from the Technical University of Munich in Germany and the University of Oxford in the UK.Using the style transfer method, cell images are decomposed into "style" (perturbation/batch representation) and "content" (cell representation), which can predict the cell's response to perturbations and remove batch effects.The article, titled "Predicting cell morphological responses to perturbations using generative modeling," was also published in Nature Communications.
In addition, in an article published by the University of Michigan team titled "MorphNet Predicts Cell Morphology from Single-Cell Gene Expression",proposed a computational method called MorphNet that can draw morphological images of cells based on their gene expression profiles.The method uses paired morphological and molecular data to train a neural network to predict the morphology of the nucleus or the entire cell based on gene expression.
In summary, promoting phenotypic drug development and biological research by observing and analyzing changes in cell states under genetic or drug perturbations has become an important topic. Regarding MorphDiff, although it still has many shortcomings, such as the challenge of predicting unseen perturbations in the face of a large number of new perturbations outside the training data, it is clear that MorphDiff is constantly improving on the basis of its predecessors, surpassing them in practicality, generalization, ease of use, and scalability.
Get high-quality papers and in-depth interpretation articles in the field of AI4S from 2023 to 2024 with one click⬇️