Command Palette
Search for a command to run...
A Hundred Universities Have Launched the world's Largest multi-cohort Proteogenomics Study, Unlocking disease-causing Genes and Repurposing Existing Drugs Based on Data From Nearly 80,000 participants.

The human genome is like a complete instruction manual for life, recording all genetic information such as appearance, height, physique, and disease risk. However, deciphering this manual isn't a straightforward process; various "unexpected events" can occur, including pathogenic mutations that predispose individuals to certain diseases. Even more challenging is...Most pathogenic variants are located in non-coding regions of the genome that do not directly encode proteins.This "black box" mechanism, which doesn't specify which gene causes the disease or through what mechanism, severely limits our ability to deduce the pathogenic genes and mechanisms. And as the direct executors that bring gene functions to life,The thousands of proteins circulating in human blood are key to unlocking black-box mechanisms and connecting non-coding variations with disease-related mechanisms.
Currently, proteogenomics research has made significant progress in clinical pathogenesis and potential drug targets, but there are still limitations to its systematic and large-scale application in human biology. First, past research has almost entirely focused on proximal cis-acting variants (i.e., cis-protein quantitative trait loci, cis-pQTLs).Non-coding variations may be located in regulatory regions, thus directly affecting multiple neighboring coding genes.It can also indirectly regulate proteins encoded by genes at other locations in the genome from a distance; secondly, past research on the multi-gene genetic structure of protein biomarkers affecting disease diagnosis and prognosis is still insufficient; finally, to stably and generally identify protein quantitative trait loci, repeated validation in different populations is required.Currently, very few human validation studies of this kind are being conducted in the field of broad-spectrum proteomics.
In view of this,A team from over a hundred universities and research institutions, including Queen Mary University of London and the University of Cambridge, has published the world's largest multi-cohort proteogenomics study to date.Based on a large-scale meta-analysis of proteoglycemic genomes covering 38 independent research cohorts and a total of 78,664 subjects, 24,738 quantitative trait sites of proteins were systematically identified and associated with 1,116 circulating proteins, comprehensively revealing the extensive proximity and distance genetic regulatory features at the protein level.
Machine learning was used to further analyze key pathways, cell types, and tissue origins regulating the abundance of circulating proteins, clarifying the core role of N-glycosylation in the protein regulatory network. Furthermore, distinguishing between cis and trans-regulatory differences in proteins effectively elucidates the intrinsic mechanisms of different biological phenotypes, providing evidence for screening potential protein drug targets for certain diseases. Further, triangulation analysis of trans sites uncovered deeper evidence for "drug repurposing."
The related research findings, titled "Multi-cohort proteogenomic analyses reveal genetic effects across the proteome and diseasome," were published in Cell.
Research highlights:
* The largest multi-cohort proteogenomics study to date, encompassing 38 independent study cohorts and involving a total of 78,664 participants.
* Identified 24,738 quantitative trait loci of proteins and associated them with 1,116 circulating proteins, comprehensively revealing a wide range of proximity and long-distance genetic regulatory features at the protein level.
* This study systematically elucidates the regulatory mechanisms of circulating proteins at the genetic level, providing important theoretical basis and data resources for understanding the molecular mechanisms of human diseases, identifying innovative therapeutic targets, and conducting drug repositioning research.

Paper address:
https://www.cell.com/cell/fulltext/S0092-8674(26)00385-5
Largest-scale core data: 38 international cohorts, nearly 80,000 participants
This study represents the world's largest multi-cohort proteogenomic meta-analysis.Integrating 38 international cohorts, covering 78,664 participants of European descent,Based on the analysis of 1,161 blood protein targets using Olink high-throughput proteomics technology, 24,738 finely mapped pQTLs (including 5,040 cis-pQTLs and 19,698 trans-pQTLs) were identified, and genetic regulatory data for 1,116 effective proteins were obtained.
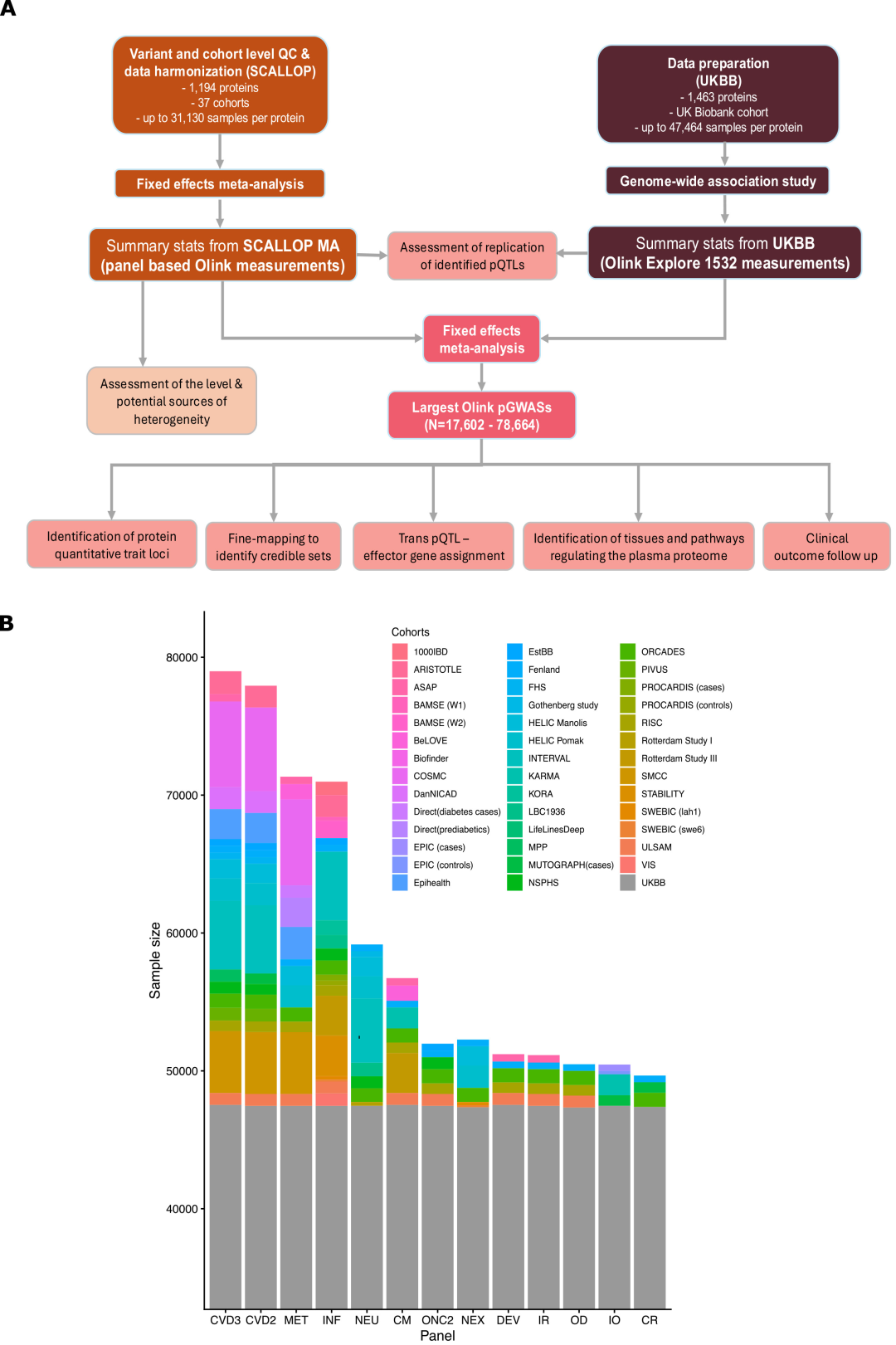
SCALLOP meta-analyses:This data includes whole-genome statistics from 37 cohorts and 1,194 blood protein targets, with the majority of participants being of European descent. Antibody-based proteomics assays were performed using at least one of 13 Target-96 assay panels provided by Olink, each capable of detecting 92 protein targets covering cardiovascular, immune, inflammatory, neurological, and metabolic fields.
UK Biobank (UKBB):The study included 48,017 participants of European descent. For this data, the proteomics measurements used in the study were generated via the Olink Explore 1536 platform, also employing antibody-based techniques to measure 1,463 protein targets.
Staged Machine Learning Classifier
The core objective of this study, employing machine learning models, is to systematically, accurately, and on a large scale assign "effect genes" to all trans-pQTLs located outside the major histocompatibility complex (MHC) region. This addresses the long-standing challenge of locating effector genes in distant genomic regions to protein quantitative trait loci associated with blood protein levels. To this end,Inspired by the ProGeM architecture, researchers have built a staged machine learning classifier.
First, regarding the sources of features and annotations, the researchers integrated multidimensional biological and genomic annotations for each genetic variant or its alternative variants (r² > 0.6). The variant-level annotations included the distance between the variant and the genome within a 1 Mb base window and the potential functional impact inferred based on the variant effect prediction (VEP) tool.
Simultaneously, gene-level annotation was performed for each gene within a 1 Mb base window, including obtaining relevant evidence of QTL colocalization based on GTEx v8 protein abundance-gene expression, rare variant load association, using the OmnipathR version 3.10.1 package to review literature and determine whether there are ligand-receptor/protein complexes corresponding to cis proteins encoded by trans genes, and determining whether related genes participate in the same biological pathway based on KEGG/REACTOME annotation information.
Then, we proceed to build the training set needed for the machine learning model, but due to the lack of widely used gold standard variants for gene allocation,Researchers used prior knowledge of biology and genomics to obtain three partially independent sets of "presumptive true positives (PTPs)".To avoid bias, only one cis protein was retained within each PTP set, and other genes within a 1 Mb window were considered negative samples. Specifically, this included trans genes encoding ligand-receptor pairs or forming high-confidence protein complexes with cis proteins (n = 540), sentinel trans-pQTLs mapped to functional variants (n = 1747), and trans genes with significant rare variant loads (n = 1049). The training and test sets were then divided into 7:3 ratios based on genomic regions, and the results were repeated 10 times to ensure stability.
Furthermore, regarding the model architecture and training process, the model algorithm in this study adopts the Random Forest classifier. By inputting 10 training sets, repeated 3-fold cross-validation is performed, combined with a subsampling strategy, thereby handling the problem of imbalanced datasets during training.Model training was implemented using the R language caret v6.0.94 toolkit, and then the best-performing random forest model in each training set was selected through Kappa score evaluation.
Then, using 10 random forest classifiers corresponding to each hypothetical true positive dataset, all candidate effector genes of trans-pQTL were scored one by one. The median score of the 10 classifiers for the same hypothetical true positive dataset was first taken, and then the three sets of predicted scores were summed. Simultaneously, when constructing the classification model for each hypothetical true positive dataset, the feature variables used to define true positive samples were removed.
Ultimately, all three classification models demonstrated stable and reliable performance, with median Kappa coefficients ranging from 0.54 to 0.57.
Deciphering the pathogenesis mechanism provides genetic evidence for drug development and repurposing of existing drugs.
This study, based on 38 international cohorts and covering 78,664 participants, conducted a multi-cohort protein genomic meta-analysis targeting 1,161 blood protein targets, systematically elucidating the genetic regulatory patterns of circulating protein levels and their association with disease.
pQTL Identification and Characteristics
The study identified 14,690 regional sentinel variants, and Bayesian fine mapping yielded 24,738 independent and reliable variant sets, encompassing 5,040 cis-pQTLs and 19,698 trans-pQTLs, covering 1,116 protein targets. Among these, cis-pQTLs were present in the protein at 87.1%, and trans-pQTLs were present in the protein at 94.1%; the cis-pQTLs at 82.3% and the trans-pQTLs at 83.3% were high-confidence sites, containing 278 newly discovered cis-pQTLs and 4,013 trans-pQTLs. Furthermore, in the non-European ancestry cohort, the effect sizes of the identified sites showed a moderate correlation with the European cohort (r = 0.6).This verifies the cross-population robustness of the results.
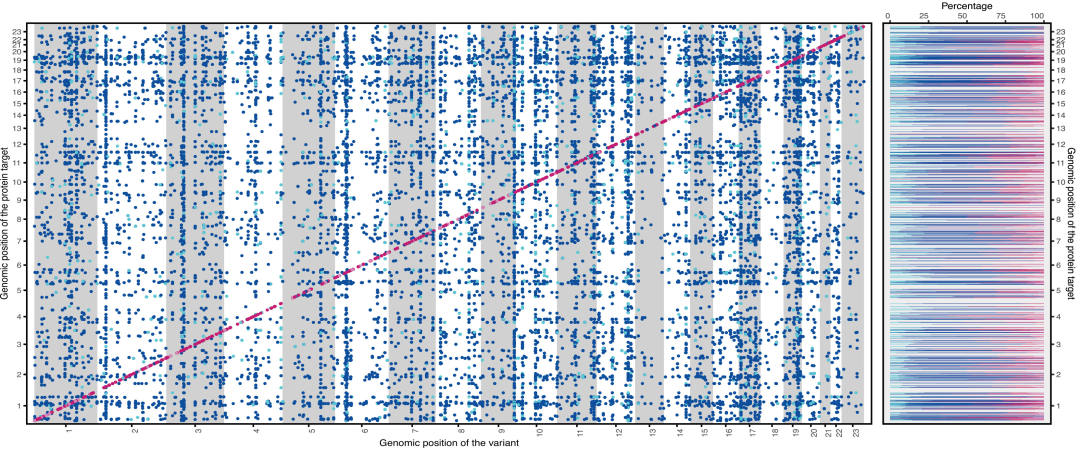
Finely located protein quantitative trait loci in SCALLOP and UKBB meta-analyses
Furthermore, there are significant differences in the explanatory power of genetic loci for variations in blood protein levels. cis-pQTLs explain an average of 8.41 TP3T of protein variation, significantly higher than trans-pQTLs. However, proteins such as ICAM2 and FUCA1 are mainly regulated by trans-pQTLs, with explanatory powers of 52.71 TP3T and 68.41 TP3T, respectively, while cis-pQTLs explain only 0.31 TP3T and 6.31 TP3T.
Furthermore, further observation of 261 protein targets revealed no significant linear correlation between the explanatory power of their pQTL variations and polygenic heritability, suggesting that this study may have reached near saturation in the identification of pQTLs for these proteins.
Characteristics of protein targets under gene regulation
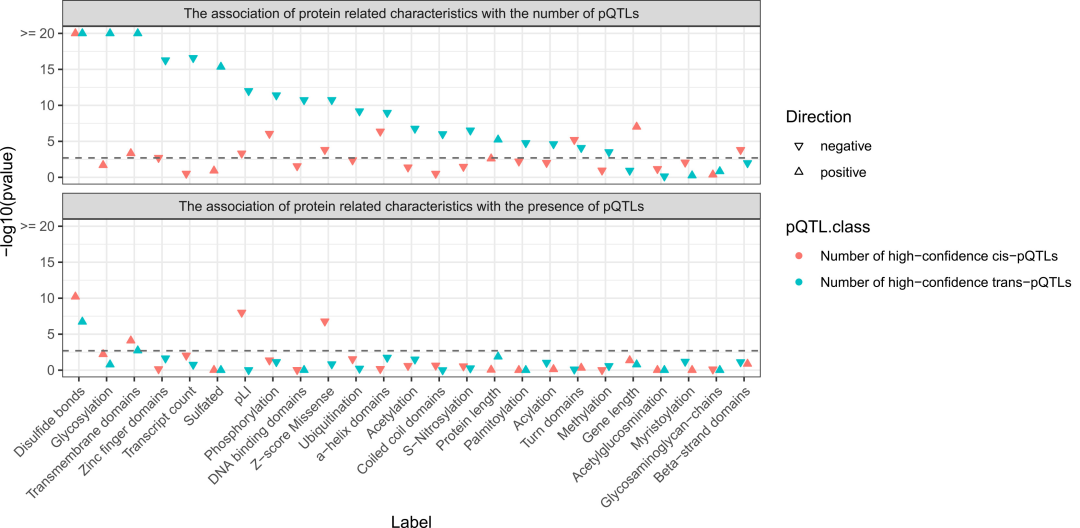
Protein characteristics related to the presence and quantity of pQTLs based on a zero-inflated Poisson regression model.
Proteins containing disulfide bonds and transmembrane domains have significantly more pQTLs, which may explain why these proteins are more easily genetically regulated; while the functional constraint strength of protein-coding genes is significantly negatively correlated with the number of cis-pQTLs.
Proteins with a high number of trans-pQTLs are significantly enriched with secretory protein features such as glycosylation and sulfation, but lack intracellular protein features such as zinc finger structures and DNA-binding domains, indicating that the long-range genetic regulation of circulating proteins is closely related to the secretory pathway.
Analysis of trans-pQTL effector genes and regulatory pathways
Based on the integration of prior biological knowledge into the machine learning framework, at least one effector gene with moderate confidence was identified for more than half of the trans-pQTLs (n = 11,261), of which 1,534 were high-confidence assignments; for two-thirds of the sites (n = 13,881), the distribution of candidate scores across genes indicated that a single causal gene was the most likely pathogenic gene.
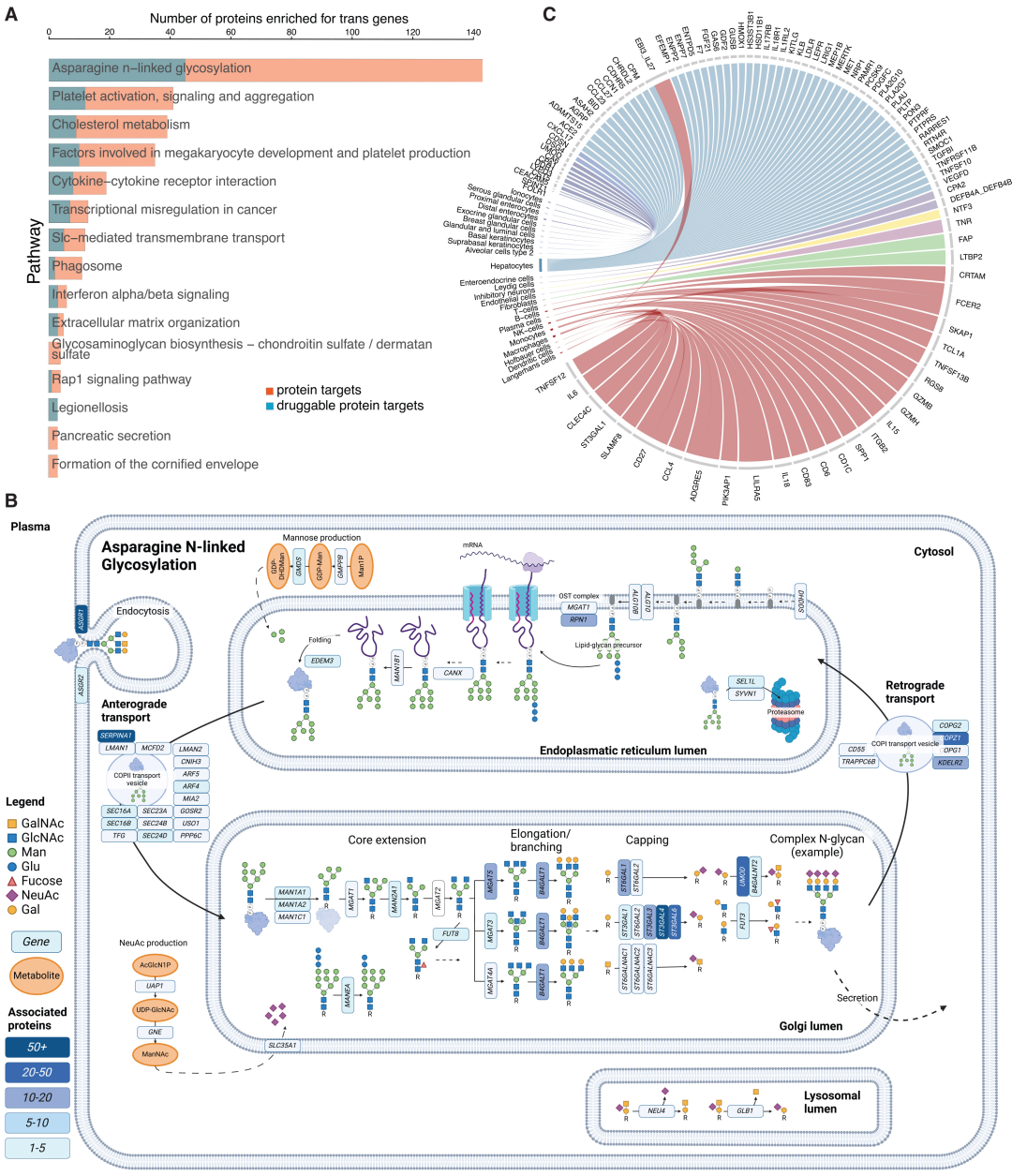
Functional enrichment analysis showed that trans-effect genes were significantly enriched in the asparagine N-glycosylation pathway (involving 143 protein targets) and platelet activation (involving 41 protein targets), among others.N-glycosylation is the most common and core regulatory pathway.
Cell and tissue enrichment results showed that trans-effect genes were highly expressed mainly in hepatocytes, natural killer cells, endothelial cells, and type II alveolar cells, revealing that the liver and immune cells are key sites for the remote regulation of circulating proteins. 44 protein-tissue pairs and 76 protein-cell type pairs were of non-classical secretory origin, confirming the important role of inter-organ communication in protein homeostasis regulation.
pleiotropic effects at the molecular and phenotypic levels
Of all the identified independent pQTLs, 43.41 TP3T exhibited pleiotropic effects, with trans-pQTLs showing significantly higher pleiotropic effects than cis-pQTLs. Subsequent studies categorized pleiotropic genetic variations into three types: "molecular pleiotropic," "phenotypic pleiotropic," and "non-specific pleiotropic." More than half (332 out of 533) showed phenotypic pleiotropic effects.In particular, its expression was enhanced by 2-fold in hepatocytes, and it preferentially regulated the target protein through protein complexes, ligand-receptor interactions, and pathway synergy.
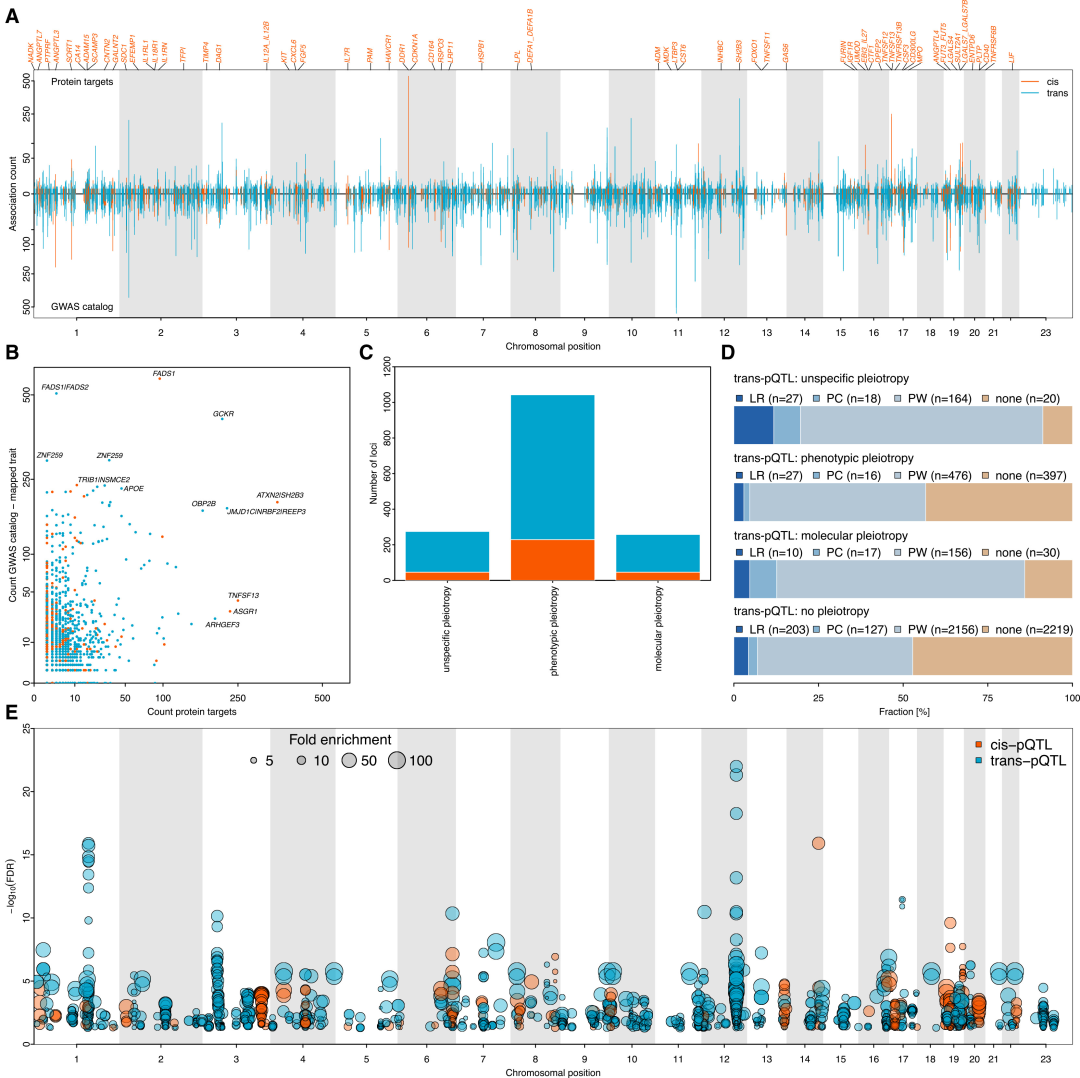
285 pleiotropic pQTLs overlap with disease GWAS sites, and their associated proteins are significantly enriched in specific pathways, providing new clues for elucidating the mechanisms of disease GWAS sites.
Differences in disease phenotypes under cis and trans regulation
Researchers combined 300 identified cis-pQTL-driven protein-disease associations with over 700 disease datasets from the FinnGen project. Only 73 of these associations yielded statistical co-localization analyses that captured both Mendelian randomization (MR) and genetic risk signals.This suggests that complementary evidence is needed when prioritizing potential disease-causing genes.
Of the 115 evaluable associations, 31 showed consistent cis- and trans-regulatory effects, 41 had no supporting evidence, and 14 showed opposite effects, indicating a significant difference in the impact of cis-proximal regulation and trans-distal regulation on disease phenotype.
Protein-disease association analysis in genetic inference and observational studies
This study integrates observational data from 52,164 participants in the UKBB study and genetic data from over 1.29 million individuals in the PanBio database, covering 517 diseases. Of the 193 high-confidence genetic associations, only 52 were consistently supported by observational studies; and of the 52,887 significant observational associations, only 0.061 TP3T received genetic evidence. Notably, blood furin protein is one of the few targets consistently associated with hypertension, myocardial infarction, and atrial fibrillation in both genetic and observational studies, revealing its potential value for drug development.
trans-pQTL guides disease biomarker discovery and drug retargeting
More than 901 TP3T disease protein biomarkers (280 out of 307 diseases) were significantly enriched in trans-pQTL-associated proteins, confirming that trans-regulation is the core genetic basis of disease protein biomarkers. The study found that the TYK2 gene missense mutation rs34536443, as a trans-pQTL, regulates multiple inflammatory proteins such as BST2 and CXCL9/10/11. Elevated levels of these proteins are associated with an increased risk of rheumatoid arthritis, psoriasis, and autoimmune thyroiditis, providing genetic evidence for the repositioning of TYK2 inhibitors for autoimmune diseases.
Conclusion
This study, based on the world's largest multi-cohort proteogenomic analysis, systematically elucidated the genetic regulatory patterns of the human circulating proteome. It broke through the limitations of previous studies that only focused on cis-regulation, and for the first time comprehensively revealed the key role of trans-genetic regulation in the regulation of circulating protein abundance at a large sample level. Furthermore, it used machine learning to precisely locate effector genes, clarifying core pathways such as N-linked glycosylation and platelet biology, as well as key regulatory sites such as the liver and immune cells.
Although this study has some limitations, such as the fact that proteomics technology only covers some subtypes and post-translational modifications of circulating proteins, and that the main population is of European descent and needs to be expanded to more ethnic groups, it still establishes a complete framework linking non-coding genetic variations, circulating proteins, and disease mechanisms. This not only provides a new perspective for elucidating the molecular mechanisms of complex diseases, but also anchors key targets such as plasma furin and TYK2 through genetic evidence, providing highly credible genetic evidence for innovative drug development and drug repurposing, and promoting a crucial step in the translation of proteogenomics from basic discovery to clinical application.








