Command Palette
Search for a command to run...
TACK: A statistical evaluation of degradation activity on a novel TArgeting Chimeras Knowledge dataset
TACK: A statistical evaluation of degradation activity on a novel TArgeting Chimeras Knowledge dataset
Stefano Ribes Nils Dunlop Rocío Mercado
Abstract
Proteolysis-targeting chimeras (PROTACs) represent a promising therapeutic modality that induces targeted protein degradation by hijacking the ubiquitin-proteasome system. However, rational PROTAC design remains challenging due to the complex interplay between molecular structure, target proteins, E3 ligases, and the cellular context. We present TACK, a statistical evaluation of degradation activity on a novel TArgeting Chimeras Knowledge dataset of 3,514 PROTACs and 6,561 degradation endpoints aggregated from three major repositories with standardized molecular representations, protein annotations, and experimental conditions. Using scaffold-based 5×5 cross-validation, we perform a rigorous statistical comparison of three machine learning methods to predict PROTAC degradation activity across three tasks: DC50 and Dmax regression, and binary activity classification. Feature ablation demonstrates that cellular context features and simple protein representations rival complex ESM protein embeddings, highlighting the importance of feature engineering over architectural sophistication. Models trained on the best performing features show that potency (pDC50, R2 = 0.66) is substantially more predictable than maximum degradation (Dmax, R2 = 0.36).
One-sentence Summary
TACK is a statistical evaluation of degradation activity on a novel TArgeting Chimeras Knowledge dataset aggregating 3,514 PROTACs and 6,561 degradation endpoints from three major repositories, utilizing scaffold-based 5×5 cross-validation to compare three machine learning methods across DC50, Dmax, and binary classification tasks where feature ablation demonstrates that cellular context features rival complex ESM protein embeddings, highlighting the importance of feature engineering over architectural sophistication.
Key Contributions
- The paper introduces TACK, a novel dataset comprising 3,514 PROTACs and 6,561 degradation endpoints aggregated from three major repositories with standardized molecular representations. An accompanying open-source ensemble model serves as a fast predictor supporting both classification and regression tasks.
- A rigorous statistical comparison of three machine learning methods is performed using scaffold-based 5×5 cross-validation to predict degradation activity across regression and classification tasks. Experimental results indicate that potency is substantially more predictable than maximum degradation, achieving an R2 of 0.66 for pDC50 versus 0.36 for Dmax.
- Feature ablation demonstrates that cellular context features and simple protein representations rival complex ESM protein embeddings in predictive performance. This finding highlights the importance of feature engineering over architectural sophistication for predicting PROTAC degradation activity.
Introduction
Proteolysis-targeting chimeras (PROTACs) offer a transformative approach to drug discovery by degrading disease-relevant proteins, yet rational design remains difficult due to complex molecular interactions and fragmented data. Prior machine learning efforts are limited by inconsistent activity metrics across repositories and evaluation protocols that often overestimate performance on novel chemical structures. The authors introduce TACK, a standardized dataset of 3,514 PROTACs with harmonized annotations to enable rigorous benchmarking. Their analysis reveals that classical machine learning methods significantly outperform specialized graph neural networks when trained on cellular context features, while an ensemble approach provides uncertainty estimates for experimental prioritization.
Dataset
-
Dataset Composition and Sources
- The authors aggregate data from three open repositories: PROTAC-DB 3.0, PROTACpedia, and TPDdb.
- Initial raw data exceeds 29,000 entries but rigorous curation reduces this to 3,514 unique PROTACs.
- The final collection comprises 6,561 experimental endpoints split between potency (DC50) and efficacy (Dmax) measurements.
-
Filtering and Standardization Rules
- Molecules undergo SMILES canonicalization using RDKit while protein names map to UniProt identifiers.
- Concentration values convert to nanomolar units and inequality operators store separately for filtering during evaluation.
- Categorical activity grades and rows containing multiple targets exclude to ensure data consistency.
- Cell lines validate against the Cellosaurus database and assay descriptions standardize to common formats.
-
Data Splits and Model Usage
- A hold-out test set represents approximately 10% of the data selected by structural dissimilarity scores.
- Remaining data uses scaffold-based clustering with Murko scaffolds for cross-validation to prevent chemical structure leakage.
- Regression tasks apply quantile transformation fitted exclusively on the training set.
- Binary classification labels compounds as active if Dmax exceeds 80% and DC50 stays below 100 nM.
-
Feature Engineering and Metadata Processing
- Protein features derive from amino acid sequences while PROTAC features use molecular fingerprints.
- Experimental conditions encode cell lines via text embeddings and assay types using one-hot encoding.
- Missing treatment times impute with the training set mean and missing cell lines embed as an unknown string.
- The curated dataset and curation pipeline are publicly available via HuggingFace and GitHub.
Method
The proposed framework integrates data curation, feature extraction, model training, and rigorous evaluation to predict PROTAC degradation activity. Refer to the framework diagram below for a visual overview of the pipeline.
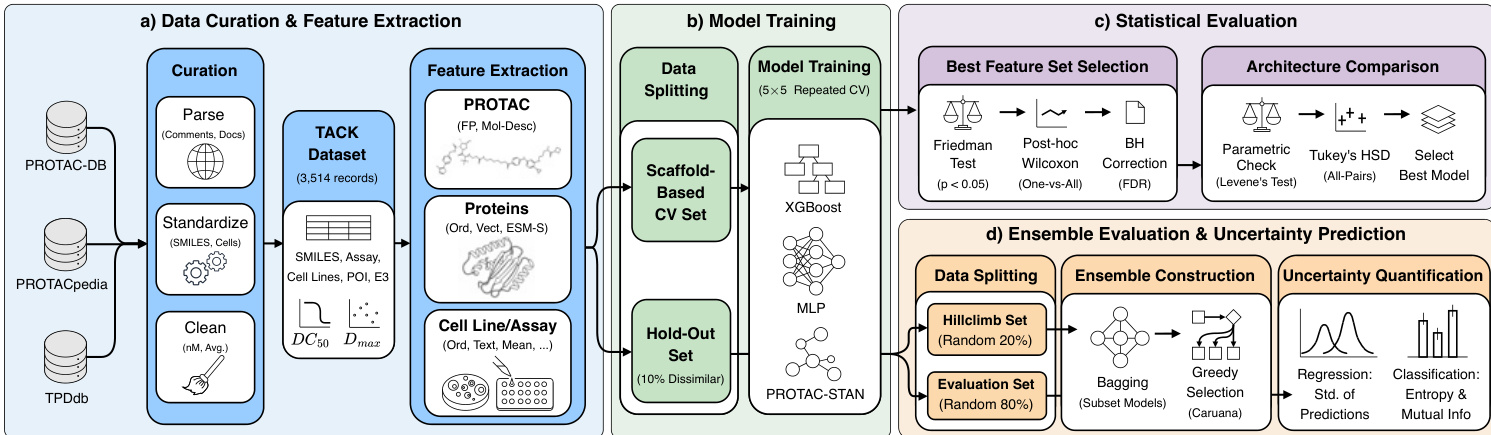
The process initiates with data curation from multiple sources, including PROTAC-DB and PROTACpedia, to construct the TACK dataset. Feature extraction is performed across three modalities: PROTAC molecules are represented using fingerprints and molecular descriptors, proteins are encoded via ESM-S embeddings, and cell line or assay data is processed into ordinal, text, or mean values.
For model training, the data is partitioned into a scaffold-based cross-validation set and a hold-out set containing 10% dissimilar compounds. The authors leverage three distinct architectures: XGBoost, a custom Multilayer Perceptron (MLP), and PROTAC-STAN. The MLP architecture is designed with flexible depth and width, utilizing hidden layers with dimensions sampled from configurations such as [256] or [512,256]. Each hidden layer incorporates optional normalization and dropout regularization, followed by a regression head with configurable depth. Weights are initialized using Kaiming initialization to account for ReLU nonlinearities, and training utilizes gradient clipping and mixed-precision training. PROTAC-STAN is a graph neural network that encodes PROTAC molecules using a two-layer edge-aware graph convolutional network and protein sequences through a fully connected adapter. A specialized Ternary Attention Network (TAN) layer models the complex three-way interactions between the protein of interest, the PROTAC, and the E3 ligase via tensor outer products.
To ensure robust predictions, the authors implement an ensemble strategy and uncertainty quantification module. The ensemble is constructed using Caruana's greedy forward selection algorithm, which iteratively builds the model set by maximizing performance on a dedicated selection subset. Bagging is applied to reduce sensitivity to selection set variability. Uncertainty estimates are derived from inter-model disagreement within the ensemble. For regression tasks, the standard deviation σ across member predictions is calculated. For classification, predictive entropy is computed and decomposed to isolate mutual information as a measure of epistemic uncertainty. Calibration is assessed by measuring the Spearman correlation between uncertainty and absolute prediction error for regression, and by reporting Expected and Maximum Calibration Error for classification. Finally, statistical evaluation involves the Friedman Test and Tukey's HSD to select the optimal feature sets and compare model architectures.
Experiment
The study utilizes a rigorous repeated 5x5 cross-validation scheme on the TACK dataset to validate feature combinations and compare XGBoost, MLP, and PROTAC-STAN architectures for PROTAC activity prediction. Statistical testing reveals that tree-based models consistently outperform deep learning approaches, with simple feature encodings often matching the performance of complex embeddings while offering greater computational efficiency. Furthermore, results indicate that while potency prediction generalizes well to hold-out sets, maximal degradation efficacy remains challenging to quantify accurately due to biological factors not captured by current molecular representations, and ensemble methods provided calibrated uncertainty estimates despite not improving predictive accuracy.
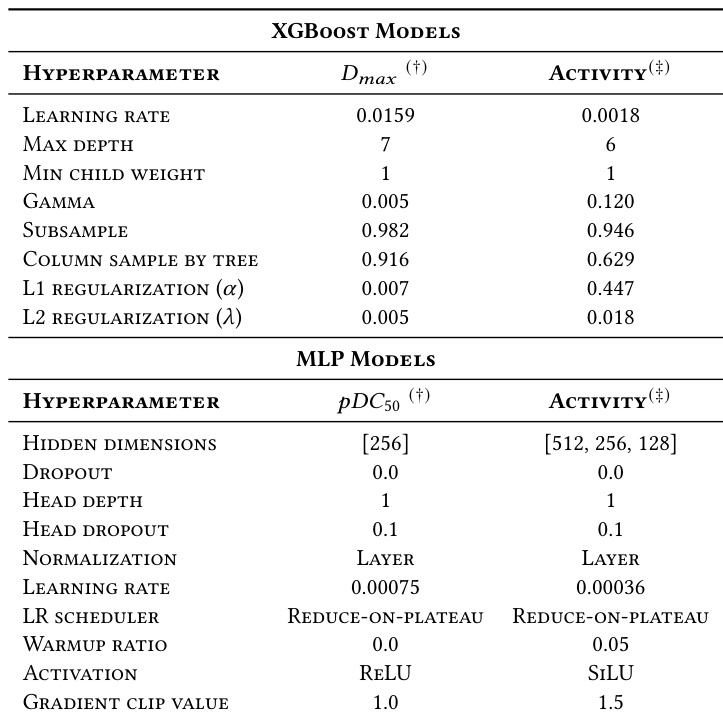
The authors present optimized hyperparameters for the best-performing XGBoost and MLP models tailored to specific prediction tasks including Dmax, pDC50, and binary Activity. The configurations reveal distinct architectural choices, such as deeper tree structures for regression tasks in XGBoost and varying hidden layer depths in MLPs to accommodate different target variables. XGBoost models for Dmax prediction use a higher learning rate and greater max depth compared to the binary Activity classification model. The MLP model for pDC50 regression employs a single hidden layer with ReLU activation, whereas the Activity model uses a deeper multi-layer architecture with SiLU activation. MLP training utilizes very low learning rates for both tasks, with the Activity model incorporating a warmup ratio and higher gradient clip values compared to the pDC50 configuration.
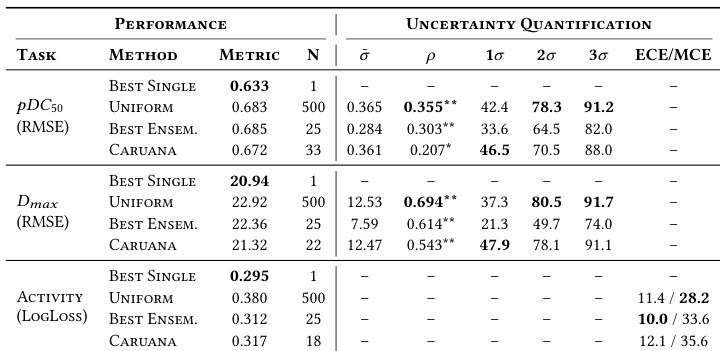
The authors compare ensemble strategies against a single best model on a held-out test set, finding that the single model consistently achieves the lowest prediction error across regression and classification tasks. While ensembles generally underperform the single best model in accuracy, they provide uncertainty estimates that correlate with prediction errors, with the uniform averaging method showing the strongest correlation for regression tasks. The study highlights a trade-off where ensembles offer better uncertainty quantification but slightly reduced predictive performance compared to the optimal single configuration. The single best model consistently achieves the lowest error metrics across all tasks, outperforming uniform and weighted ensemble approaches. Uniform averaging yields the strongest correlation between estimated uncertainty and actual prediction error for regression tasks. Ensemble methods demonstrate moderate calibration for binary classification, with the best ensemble showing the lowest calibration error.
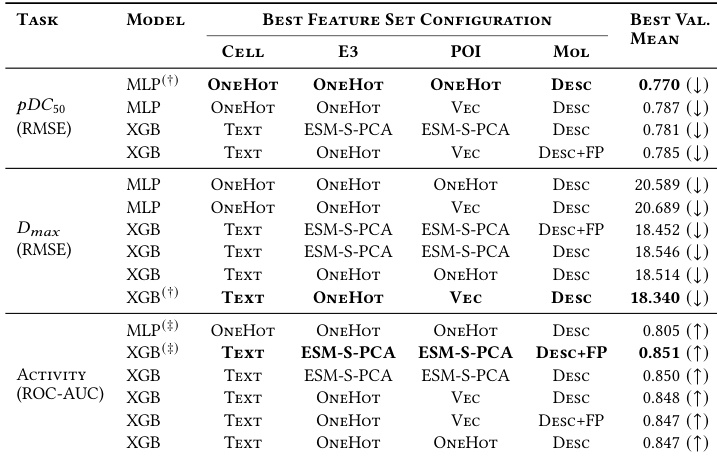
The authors compared XGBoost and MLP models across three prediction tasks, finding that XGBoost generally achieved superior performance for maximal degradation and activity classification. In contrast, the MLP model demonstrated slightly better results for potency prediction using simpler feature representations. XGBoost achieved the best performance for Dmax regression and Activity classification tasks. MLP outperformed XGBoost on the pDC50 regression task using simple OneHot encodings. XGBoost configurations frequently incorporated ESM-S-PCA embeddings, whereas MLP configurations consistently relied on OneHot encodings.
The authors evaluate optimized XGBoost and MLP configurations across multiple prediction tasks, noting that distinct architectural choices are required to handle regression and classification objectives effectively. Comparative analysis reveals that while single models consistently achieve the lowest prediction errors, ensemble strategies provide superior uncertainty quantification correlated with actual errors. Additionally, performance varies by task type, with XGBoost excelling in maximal degradation and activity classification while MLP demonstrates better results for potency prediction using simpler feature encodings.