Command Palette
Search for a command to run...
Based on 14,000 real-world Datasets, the University of Washington/Microsoft and Others Proposed GigaTIME to Create a Panoramic Atlas of the Tumor Immune microenvironment.
In the evolutionary landscape of cancer, the tumor immune microenvironment not only dominates the growth, invasion, and metastasis of cancer cells, but also profoundly influences treatment response and the patient's final prognosis. This is not a "solo performance" by cancer cells, but a highly dynamic ecosystem—immune cells, fibroblasts, endothelial cells, and other roles intertwine and interact, jointly embedding themselves in the extracellular matrix, whose structure and function have been remodeled, forming a precise and complex pathological network.
The key to deciphering this network lies in understanding the functional states and interactions of cells, and the activation levels of specific proteins are crucial "molecular codes." Traditionally,Immunohistochemistry (IHC) has become a classic tool for deciphering codes due to its ability to visually visualize protein localization.For example, PD-L1 staining has been widely used to identify the status of immune checkpoints to predict the efficacy of immunotherapy. However, IHC can only capture information about one protein at a time, making it difficult to reconstruct the true ecology of multiple protein coexistence, which constitutes a major bottleneck in a deeper understanding of the tumor-immune cell dialogue mechanism.
To overcome this limitation, multiplex immunofluorescence (mIF) technology was developed. It can simultaneously present the spatial distribution of multiple proteins on a single tissue section, fully preserving the contextual information of the tissue structure.However, this technology is expensive and has a complicated process, with staining, imaging, and analysis all being extremely time-consuming.This makes it difficult to accumulate large-scale data and hinders clinical translation.
In stark contrast, H&E-stained sections are widely available and inexpensive in clinical settings. While they cannot directly reveal protein activity, they completely preserve the overall structure of the tissue and the details of cell morphology. The hidden features within may indirectly reflect the functional state of the cells, but these subtle and complex patterns often exceed the limits of human visual perception.
In recent years, breakthroughs in artificial intelligence technology have brought new opportunities. Through pre-training on massive amounts of pathological images, AI has demonstrated powerful visual analysis and feature mining capabilities. This leads to a key question: can AI be used to "decode" protein activation information from readily available H&E images, information that previously required expensive mIF technology to capture?
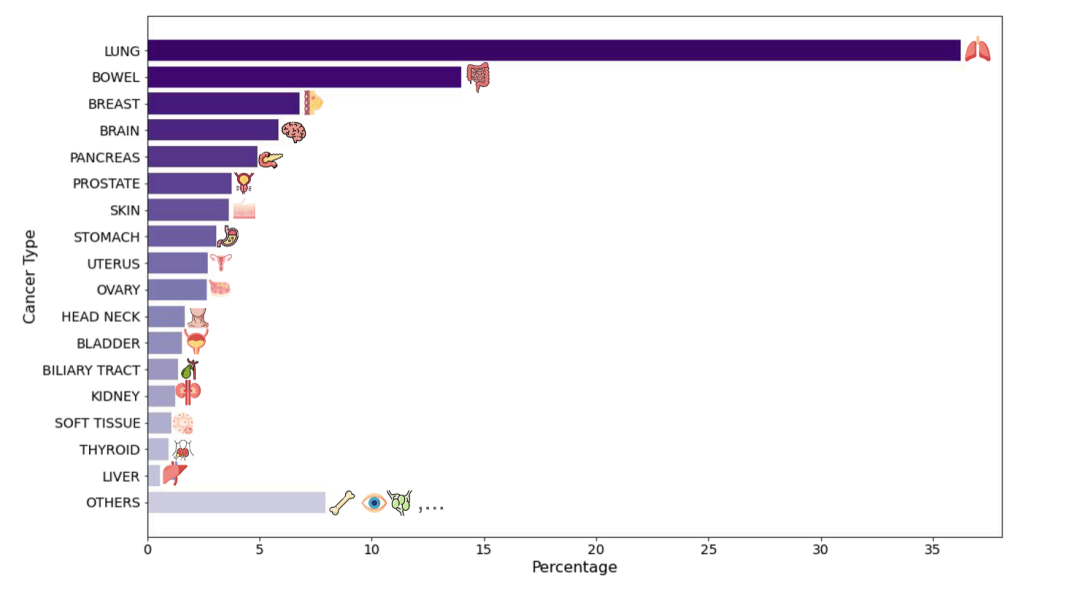
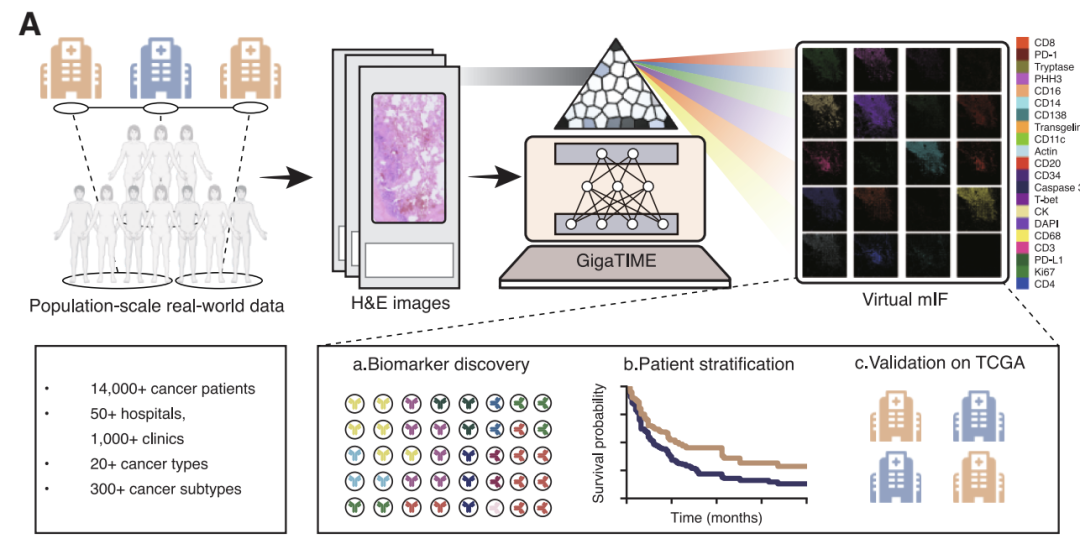
Based on this line of thinking,A research team comprised of Microsoft Research, the University of Washington, and Providence Genomics has proposed GigaTIME, a multimodal artificial intelligence framework.Leveraging advanced multimodal learning technology, it can generate virtual mIF maps from conventional H&E slices. The research team applied it to a cohort of over 14,000 cancer patients at Providence Medical Center in the United States, covering 24 cancer types and 306 subtypes, ultimately generating nearly 300,000 virtual mIF images, achieving systematic modeling of the tumor immune microenvironment in a large and diverse population.
The related research findings, titled "Multimodal AI generates virtual population for tumor microenvironment modeling," have been published in Cell.
Research highlights:
* GigaTIME uses multimodal AI to convert H&E pathology slides into spatial proteomics data, generating virtual populations containing cellular states from routine H&E slides.
* Supports large-scale clinical discovery and patient stratification, and reveals novel spatial and combinatorial protein activation patterns.

Paper address:https://www.cell.com/cell/fulltext/S0092-8674(25)01312-1
Follow our official WeChat account and reply "Multiple Immunizations" in the background to get the full PDF.
More AI frontier papers:
https://hyper.ai/papers
Datasets: Building a complete closed loop from training to application
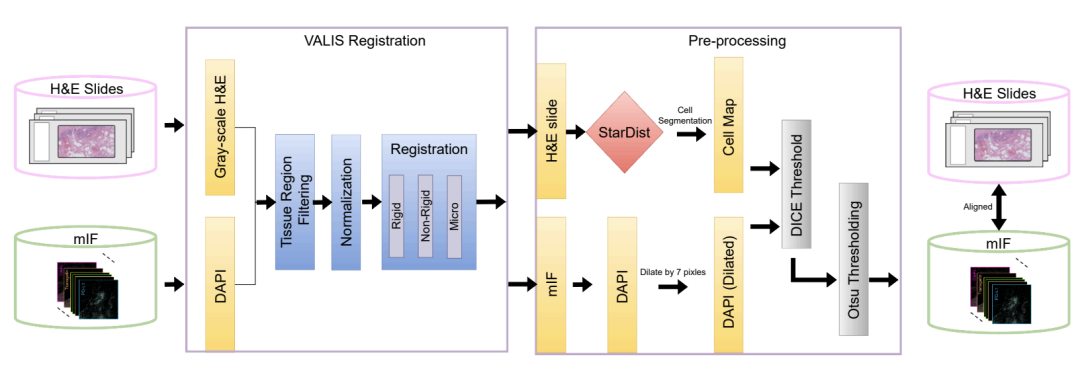
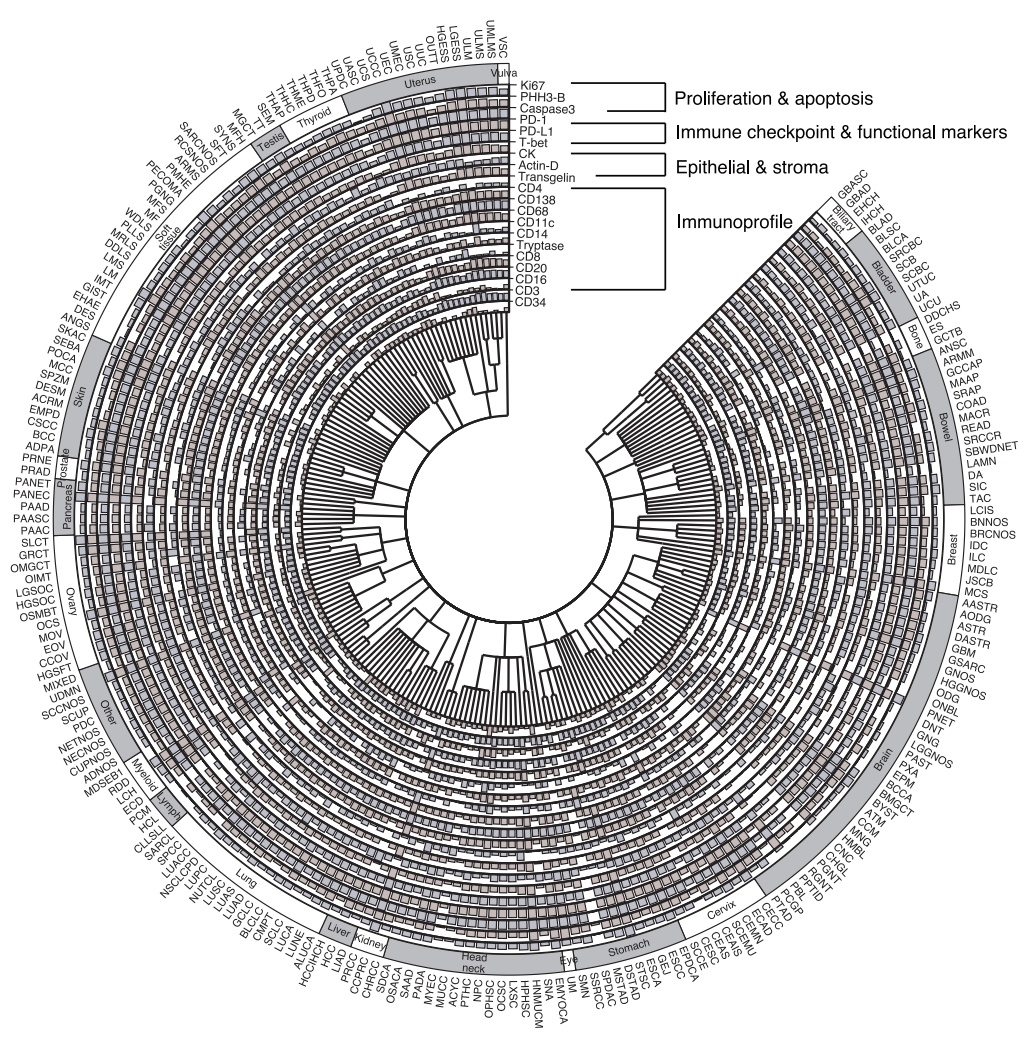
Training a model first requires addressing a fundamental contradiction: H&E staining, widely available and low-cost in clinical practice, cannot directly reveal protein activity, while mIF technology, which can reveal spatial relationships between multiple proteins, is expensive and complex, making large-scale implementation difficult. To construct an AI model that connects these two imaging techniques,The research team used the COMET platform to collect 441 mIF images from 21 H&E stained sections.As shown in the figure below, these images cover a total of 21 key biomarkers, ranging from nuclear proteins such as DAPI and PHH3, surface proteins such as CD4 and CD11c, to cytoplasmic proteins such as CD68. They provide important evidence for analyzing the composition, functional status, and activity of immune cells in the tumor microenvironment.

After obtaining the paired images, the greater challenge lies in extracting high-quality training data from them. To this end, as shown in the figure below, the team designed a rigorous processing workflow: First, the VALIS tool is used to precisely align the H&E image and the mIF image at the pixel level; then, the StarDist algorithm is used to identify and segment each cell in the image; finally, the image region with the highest registration quality is selected based on the Dice coefficient.
Through layers of quality controlThe team selected 10 million high-quality cells from the initial data containing 40 million cells and divided them into training set, validation set and independent test set.In addition, the study introduced breast cancer and brain cancer samples from tissue microarrays as an external validation set. These samples differed significantly from the training data in tissue structure and morphology—they appeared as small cylindrical tissue blocks separated by blank areas, rather than the large, continuous tissue slices in the training data, thus effectively testing the model's generalization ability when faced with new sample types and unseen cancer types.
At the model application level, the study constructed two large-scale and complementary virtual population cohorts.The first cohort comes from the clinical network of Providence Health, a US-based healthcare group, and includes H&E slices from 14,256 cancer patients across 51 hospitals and over 1,000 clinics. It covers 24 major cancer types and 306 subtypes, and integrates rich clinical information such as genomic biomarkers, pathological staging, and survival follow-up. The unique value of this dataset lies in its real-world characteristics: a highly diverse patient population spanning a complete spectrum of disease stages from early to late stages, realistically reflecting the complexities of clinical practice.
The second cohort was taken from the Cancer Genome Atlas (TCGA) public database.The study included 10,200 H&E slides, primarily from early-stage, untreated surgical cases. These two cohorts provided a stark contrast in patient origin, disease stage, and clinical context. This differentiated design offered excellent conditions for validating the model's reliability and generalizability: consistent and robust biological conclusions across such diverse datasets strongly suggest its broad clinical potential.
GigaTIME: Building Intelligent Bridges of Form and Function
The GigaTIME model directly addresses a key bottleneck in tumor immune microenvironment research: the high-cost, low-throughput mIF technology is difficult to popularize, while routine clinical H&E staining images cannot directly reflect protein functional activity. This model uses artificial intelligence to learn and generate virtual mIF images from H&E images, thus providing a feasible path for low-cost, systematic analysis of the tumor immune microenvironment at the population scale.
The model employs a carefully designed patchwork encoder-decoder framework, the core of which is built upon a nested U-shaped network.The advantage of this architecture lies in its ability to simultaneously capture both subtle local features and global organizational structure of an image. Specifically, the encoder part of the network extracts multi-level feature representations from the input 256×256 pixel H&E image patch through convolution and downsampling operations; the decoder part then reconstructs these abstract features into a virtual mIF image with spatial resolution through upsampling and feature fusion. This design allows the model to focus on both the fine morphology of individual cells and the organizational patterns of cell populations.
At the output level, the model's design reflects a deep consideration of biological issues.For each of the 21 preset protein channels, GigaTIME performs binary classification prediction for every pixel in the input image.The system determines whether a specific protein is activated at a given location, ultimately generating a pixel-level protein activity map. These local predictions can be seamlessly stitched together to reconstruct a virtual mIF image of the entire tissue slice. This supports the calculation of various quantitative indicators, such as the activation density and spatial distribution pattern of specific proteins in the tumor region, providing a solid data foundation for subsequent high-throughput analysis and clinical correlation studies.
To ensure effective model learning, the training strategy has been systematically optimized.The loss function cleverly combines Dice loss and binary cross-entropy loss: the former focuses on ensuring the overall consistency of the predicted active region and the ground truth region in terms of spatial contour, while the latter focuses on improving the classification accuracy of each pixel. The synergy of the two ensures both accurate reconstruction of the global spatial pattern and reliability at the detail level. The model was trained for 250 epochs on 8 NVIDIA A100 GPUs with a batch size of 16 and a learning rate of 0.0001. All key hyperparameters were determined through system debugging based on the validation set results.
It is particularly important to emphasize that the success of the model depends heavily on high-quality training data.The research team employed rigorous image registration, cell segmentation, and quality control procedures.10 million high-quality cells were selected from the massive initial data for training, ensuring that the model learns robust, reliable and biologically meaningful cross-modal mappings, rather than superficial statistical regularities or noisy patterns.
Large-scale findings based on nearly 300,000 virtual images: GigaTIME reveals 1,234 clinical associations
To comprehensively evaluate the performance and value of GigaTIME, the research team designed a systematic evaluation scheme from two dimensions: technology validation and clinical findings.
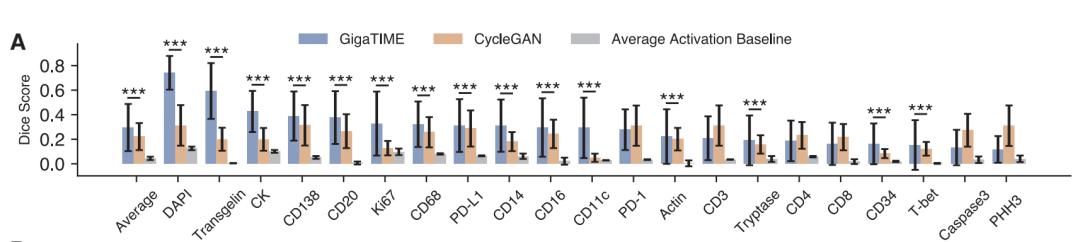
In terms of technical verification,The study evaluated the model's image conversion capabilities at three levels: pixel, cell, and slice.At the pixel level, GigaTIME significantly outperforms the baseline model CycleGAN on 15 out of 21 protein channels. For example, on the DAPI channel, GigaTIME achieves a Dice coefficient of 0.72, far exceeding the 0.12 of the simple statistical baseline.
At the cellular level,GigaTIME achieved a correlation of 0.59 on the DAPI channel, while CycleGAN only reached 0.03, approaching a random level.
At the slice levelGigaTIME's DAPI channel correlation coefficient is as high as 0.98, with an average of 0.56 across all channels, while CycleGAN's is close to 0. These results demonstrate that supervised training based on high-quality paired data is crucial for accurate cross-modal conversion.
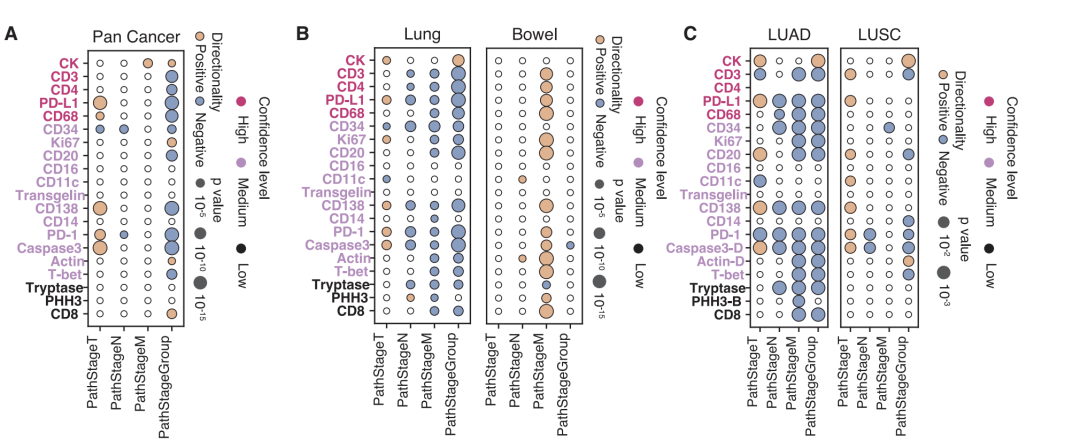
In terms of clinical findings, the study utilized nearly 300,000 virtual mIF images from 14,256 patients.The association between virtual protein expression and 20 clinical biomarkers was systematically analyzed.After rigorous statistical testing and multiple corrections, a total of 1,234 significant associations were identified, distributed across three levels: pan-cancer, cancer type, and cancer subtype.
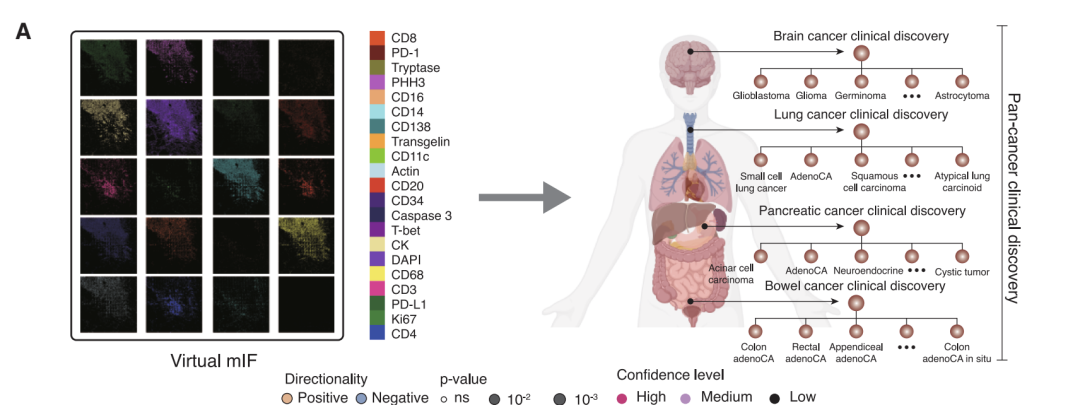
Among the 175 associations identified in the pan-cancer analysis, high tumor mutational burden and high microsatellite instability were significantly associated with enhanced activation of multiple immune infiltration markers (CD138, CD20, CD68, and CD4), consistent with an antigen-driven immune activation mechanism. New clues were also discovered: KMT2D mutations showed a strong positive correlation with immune markers, suggesting a potential promotion of immune infiltration; while KRAS mutations showed a negative correlation, reflecting an immune rejection phenotype. In specific cancer types and subtypes, the model revealed numerous specific associations. For example, the strong correlation between T-bet and TP53 mutations in brain cancer was not detected at the pan-cancer level, possibly related to the unique immune microenvironment of the central nervous system. Lung cancer subtype analysis showed that PRKDC mutations in lung adenocarcinoma were more strongly associated with immune response markers than in squamous cell carcinoma, confirming the importance of interpreting the data in conjunction with histological context.
The study also validated the value of virtual data in clinical outcomes.Analysis revealed a positive correlation between primary tumor size (T stage) and immune checkpoints and invasion markers, but this association reversed in advanced stages, suggesting that advanced tumors may be primarily driven by other immune escape mechanisms. In survival analysis, the composite features integrating all 21 pathways were superior to single-protein analyses in patient stratification, fully demonstrating the value of multi-parameter combined analysis.
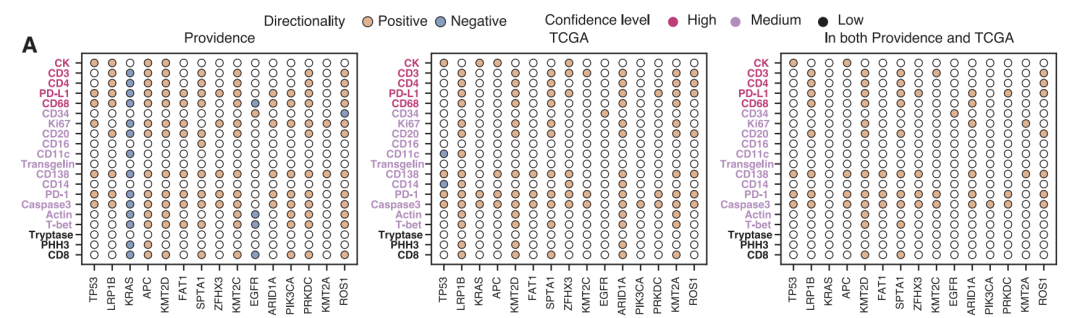
To ensure reliability, all key findings were validated in independent cohorts from TCGA. Despite significant differences in origin and clinical characteristics between the two populations, the core findings remained highly consistent (Spearman correlation coefficient of 0.88 at the cancer subtype level).The 80 commonly identified significant associations showed extremely high statistical enrichment (p<2×10⁻⁹).Meanwhile, Providence Health's virtual population showed 331 more significant associations at the pan-cancer level than TCGA, highlighting the unique value of large-scale real-world data.
Exploratory analysis also revealed the value of complex spatial patterns. Indicators such as entropy, signal-to-noise ratio, and sharpness outperformed simple activation density in 89, 63, and 79 protein-biomarker pairs, respectively. The study also discovered synergistic effects between proteins:The combination of CD138 and CD68 outperformed single proteins in predicting 20 biomarkers.Thirteen of these differences were statistically significant, suggesting that plasma cells and macrophages may work together to fight tumors through an antibody-mediated mechanism.
AI-Powered: From Virtual Protein Maps to New Frontiers in Cancer Research
Generating virtual proteomics images from routine pathology slides using AI is at the heart of innovation in digital pathology and computational biology. This direction has attracted exploration from top academic institutions worldwide and has also spurred commercial practices by biotechnology companies.
In academia,The HEX model published by Stanford University in Nature MedicineTrained on 819,000 paired image blocks, it can predict the spatial expression of 40 biomarkers, demonstrating broader protein coverage than GigaTIME. The DeepHeme system, published by the University of California, San Francisco in Science Translational Medicine, based on nearly 50,000 high-quality multicenter datasets, has achieved accurate classification of 23 types of bone marrow cells, providing a paradigm for the automation of hematological disease diagnosis.
In the industry, Reveal Biosciences is supported by the Bill & Melinda Gates Foundation.Develop a platform to extract "digital biomarkers" from pathological images.Accelerating global health research. Another approach is to reduce costs through hardware innovation, such as Micronit's microfluidic devices that significantly reduce sample and reagent consumption. Optellum's FDA-approved lung nodule diagnostic platform provides a commercial paradigm and regulatory precedent for extracting deeper features from routine data for clinical decision-making.
GigaTIME is a significant milestone in this field.This not only demonstrates the enormous potential of multimodal AI in the study of the tumor immune microenvironment, but also provides a reusable technical framework and data resources for subsequent research.Future development will depend on the combined advancement of "virtual-reality" data generation capabilities and low-cost detection technologies, ultimately bringing transformative tools for understanding tumor complexity and accelerating precision medicine.
Reference Links:
1.https://mp.weixin.qq.com/s/AsqSemP3idCbIJ7xQ3gXGg
2.https://mp.weixin.qq.com/s/umg-UrMm6Qe-R-MbLpLZOQ